Эдвардса синдром
Наша команда профессионалов ответит на ваши вопросы

«Золотым стандартом» выявления хромосомных нарушений во всем мире долгое время являлся и продолжает оставаться метод кариотипирования с дифференциальной окраской хромосом. Этот метод позволяет анализировать кариотип в целом и определять крупные (не менее 5-10 млн пар нуклеотидов) хромосомные перестройки. Однако у него существует ряд ограничений, таких как трудоемкость, длительность (1-2 недели), высокие требования к квалификации и опыту специалиста, проводящего исследование, а также, в ряде случаев, технические проблемы (недостаточное количество и качество исследуемого материала, отсутствие митозов или роста культуры).
Этих недостатков лишен метод количественной флуоресцентной полимеразной цепной реакции (КФ-ПЦР), который все более широко применяется для диагностики анеуплоидий, в том числе и синдрома Эдвардса (Рис. 1). Этот метод обладает достоверностью, сравнимой с достоверностью стандартного кариотипирования, является более быстрым, дешевым, менее требовательным к количеству и качеству материала (поскольку не связан с ростом культуры клеток) и позволяет одновременно анализировать большое число образцов. Однако метод КФ-ПЦР имеет и ограничения: в мозаичных случаях он позволяет выявлять только высокоуровневый мозаицизм (от 20%), кроме того, он не может исключить наличие более редких хромосомных нарушений, которые могут быть связаны с пороками развития плода. При проведении дородовой диагностики синдрома Эдвардса, кроме материала плода, необходимо предоставлять биологический материал матери для того, чтобы исключить возможность получения ложноотрицательного результата из-за неправильного забора плодного материала. Анализ плодного материала выполняется за три рабочих дня.
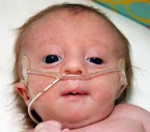
При синдроме Эдвардса отмечается выраженная задержка пренатального развития, дети рождаются с пренатальной гипотрофией (средняя масса тела при рождении составляет 2340 г). Внешние проявления синдрома Эдвардса многообразны (Рис. 2). Наиболее типичными являются задержка психомоторного развития, гипоплазия скелетной мускулатуры и подкожной жировой ткани, врожденные пороки сердца, аномалии строения лица и черепа (долихоцефалия, микрофтальмия, укорочение глазных щелей, низкое расположение ушных раковин, микрогнатия, скошенный подбородок), множественные деформации кистей и стоп, аномалии развития желудочно-кишечного тракта, мочеполовой системы и центральной нервной системы (спинно-мозговые грыжи, гипоплазия мозолистого тела и мозжечка). Продолжительность жизни детей резко снижена: 90% из них погибают до года от осложнений, обусловленных врождёнными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).
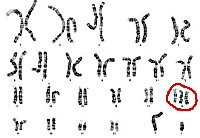
Причиной развития синдрома Эдвардса является утроение хромосомы 18. Трисомия по хромосоме 18 является частным случаем анеуплоидии– наличия в геноме набора хромосом, отличного от стандартного для данного вида и некратного ему. Трисомия хромосомы 18 обычно вызвана нерасхождением хромосом при формировании половых клеток родителя (яйцеклеток и сперматозоидов), в результате чего ребенок получает от матери или от отца лишнюю 18-ю хромосому. В этом случае все клетки организма ребёнка будут нести аномалию. В том случае, когда нерасхождение хромосом возникает при делении какой-либо клетки зародыша, наблюдается мозаичный вариант синдрома Эдвардса (10% случаев).
Риск рождения детей с синдромом Эдвардса, по разным литературным данным, не изменяется или незначительно возрастает с увеличением возраста беременной женщины.
Пренатальная диагностика синдрома Эдвардса включает в себя два этапа. На первом этапе, на сроке беременности 11-13 недель, проводится скрининг, который основывается преимущественно на биохимических показателях, поскольку на ранних сроках УЗИ не позволяет обнаружить в случае синдрома Эдвардса каких-либо грубых аномалий развития, которые могут быть выявлены лишь к 20-24 неделе. Биохимический анализ уровня определенных белков в крови беременной женщины (свободной β-субъединицы хорионического гормона человека (β-ХГЧ) и ассоциированного с беременностью плазменного протеина А (pregnancy associated plasma protein-A, РАРР-А)), с учетом ее возраста, позволяет рассчитать для нее риск рождения больного ребенка. Однако эти методы не позволяют поставить точный диагноз, и в результате проведенного скрининга лишь формируется группа риска беременных с повышенной вероятностью рождения больного синдромом Эдвардса. На втором этапе в группе риска проводится инвазивная процедура для получения плодного материала, необходимого для точного определения статуса плода. В зависимости от срока беременности это может быть биопсия ворсин хориона (8-12 недели), амниоцентез (14-18 недели) или кордоцентез (после 20-й недели). В полученных образцах ткани плода проводится определение хромосомного набора.
В Центре Молекулярной Генетики проводится диагностика синдрома Эдвардса (в том числе и пренатальная) методом КФ-ПЦР.
Синдром Эдвардса
Общие сведения
Синдром Эдвардса – количественная хромосомная аберрация, при которой имеет место частичная или полная трисомия по 18 аутосоме. Синдром получил название по имени генетика J. Edwards, подробно описавшего заболевание в 1960 г. и выделившего свыше 130 характерных для данной патологии симптоматических дефектов. Синдром Эдвардса – второе по распространенности хромосомное заболевание после синдрома Дауна; частота рождения детей с синдромом Эдвардса составляет 1:5000-7000. Примерно три четверти всех больных синдромом Эдвардса – девочки; предполагается, что большая часть беременностей плодом мужского пола заканчивается внутриутробной гибелью и самопроизвольным абортом.
Причины синдрома Эдвардса
Причиной полной трисомии служит мейотическое нерасхождение хромосом. Практически во всех случаях лишняя хромосома является материнской по происхождению. Этот вариант синдрома Эдвардса является наиболее тяжелым по своим проявлениям и неблагоприятным в плане прогноза. Возникновение мозаицизма связано с нерасхождением хромосом на ранней стадии дробления зиготы. В этом случае лишнюю хромосому будут содержать не все клетки плода, а лишь их часть. Транслокация – присоединение части 18-ой хромосомы к другой паре может произойти как в процессе созревания гамет, так и после оплодотворения. При этом клетки организма содержат две гомологичные 18-е хромосомы и ее дополнительную часть, прикрепленную к другой хромосоме.
Как и в случае с синдромом Дауна, возраст матери является наиболее значимым риск-фактором рождения ребенка с синдромом Эдвардса. В редких случаях у родителей может выявляться носительство сбалансированной транслокации.
Симптомы синдрома Эдвардса
Во время беременности наблюдается многоводие, слабая активность плода, маленькая плацента, единственная пупочная артерия. Ребенок с синдромом Эдвардса рождается с низкой массой тела (около 2170 г) и пренатальной гипотрофией при доношенной или даже переношенной беременности. У части детей определяется состояние асфиксии при рождении.
У новорожденных с синдромом Эдвардса имеются характерные фенотипические признаки, позволяющие предположить данную хромосомную патологию. В первую очередь обращает на себя внимание долихоцефалическая форма черепа с преобладанием продольного размера над поперечным, низкий лоб, выступающий затылок, микрогнатия, маленький рот, микрофтальмия. У детей с синдромом Эдвардса часто встречаются расщелины верхней губы и нёба, эпикант, птоз, экзофтальм, косоглазие, короткая шея с избыточной кожной складкой. Типичные деформации ушных раковин включают маленькие мочки, отсутствие козелков, узкие слуховые проходы, низкое расположение ушей.
При синдроме Эдвардса имеются множественные тяжелые аномалии со стороны практически всех систем организма. Врожденные пороки сердца могут быть представлены дефектами межжелудочковой и межпредсердной перегородок, коарктацией аорты, транспозицией магистральных сосудов, дисплазией клапанов, тетрадой Фалло, аномальным дренажом легочных вен, декстракардией и др. При синдроме Эдвардса может выявляться патология развития желудочно-кишечного тракта: диафрагмальные, пупочные и паховые грыжи, дивертикул Меккеля, трахеопищеводные свищи, пилоростеноз, атрезия подвздошной кишки и ануса. Наиболее частыми аномалиями мочеполовой системы у детей с синдромом Эдвардса служат подковообразная почка, гидронефроз, дивертикулы мочевого пузыря, гипоспадия и крипторхизм (у мальчиков), двурогая матка, внутриматочная перегородка и гипертрофия клитора (у девочек).
Новорожденные с синдромом Эдвардса испытывают трудности с сосанием, глотанием и дыханием, из-за чего им требуется зондовое питание или длительная ИВЛ. Дети с синдромом Эдвардса, как правило, погибают на первом году жизни из-за тяжелых врожденных пороков развития и связанных с ними осложнений (сердечно-сосудистой и дыхательной недостаточности, пневмонии, кишечной непроходимости и т. д.).
Диагностика синдрома Эдвардса
Важнейшей задачей диагностики служит антенатальное выявление синдрома Эдвардса у плода, поскольку данная патология является медицинским показанием для искусственного прерывания беременности. Заподозрить наличие синдрома Эдвардса можно в процессе УЗИ плода и допплерографии маточно-плацентарного кровотока по косвенным признакам (множественным аномалиям развития плода, агенезии пупочной артерии, малой величине плаценты, многоводию и пр.).
Наибольшую диагностическую значимость имеет стандартный пренатальный скрининг, включающий анализ крови на сывороточные маркеры: βХГЧ и PAPP на 11-13 неделе беременности; βХГЧ, альфа-фетопротеин и свободный эстриола на 20-24 неделе гестации.
При оценке степени риска рождения ребенка с синдромом Эдвардса учитываются данные биохимического и ультразвукового скрининга, срок беременности, возраст и масса тела женщины. Беременным, попадающим в группу высокого риска, предлагается проведение инвазивной дородовой диагностики (биопсии хориона, амниоцентеза, кордоцентеза) с последующим кариотипированием плода.
В случае рождения живого ребенка с синдромом Эдвардса необходимо как можно более раннее всестороннее обследование, направленное на выявление тяжелых пороков развития. Новорожденный с синдромом Эдвардса должен быть осмотрен неонатологом, детским кардиологом, детским неврологом, детским хирургом, детским ортопедом, детским урологом и др. Наиболее важными диагностическими исследованиями, которые должны быть выполнены ребенку с синдромом Эдвардса в первые часы жизни, служат эхокардиография, УЗИ органов брюшной полости и УЗИ почек.
Лечение синдрома Эдвардса
Поскольку в большинстве случаев аномалии развития оказываются несовместимыми с жизнью, лечение детей с синдромом Эдвардса сводится к оказанию симптоматической помощи, направленной на поддержание физиологических функций, продление жизни и улучшение ее качества. Хирургическая коррекция врожденных пороков, как правило, является рискованной и неоправданной.
Поскольку дети с синдромом Эдвардса ослаблены и подвержены частой заболеваемости инфекциями мочевыводящих путей, средним отитом, конъюнктивитом, синуситами, пневмониями и пр., они нуждаются в тщательно организованном уходе, полноценном питании, регулярном наблюдении со стороны педиатра.
Прогноз и профилактика синдрома Эдвардса
Во всех случаях прогноз при синдроме Эдвардса крайне неблагоприятный: в среднем мальчики живут 2-3 месяца, девочки – 10 месяцев. До 1 года доживает лишь 10% больных, до 10 лет – не более 1%. Относительно благоприятные шансы в отношении выживания имеют дети с мозаичной формой синдрома Эдвардса.
Риск рождения ребенка с синдром Эдвардса теоретически существует в любой супружеской паре; известно, что такая вероятность выше у возрастных родителей (для женщин старше 45 лет – 0,7%). С целью своевременного выявления хромосомной патологии у плода не следует пренебрегать антенатальным скринингом, входящим в программу введения беременности.
Синдром Эдвардса

Лечением данного заболевания занимается генетик.
МКБ-10: Q91 — Синдром Эдвардса и синдром Патау.
Синдром Эдвардса получил название по фамилии ученого Джона Эдвардса, впервые описавшего трисомию. Синдром Эдвардса представляет собой хромосомную патологию, которая характеризуется наличием дополнительной хромосомы (трисомия) в 18 паре. Это тяжелая врожденная патология, вторая по частоте после синдрома Дауна, характеризующаяся тяжелыми аномалиями в развитии внутренних органов с неблагоприятным прогнозом. Согласно статистическим данным, частота болезни во всем мире находится в пределах 0,015—0,02%. На три-четыре больных девочки приходится один мальчик.

Причины синдрома Эдвардса
Так как синдром Эдвардса — это хромосомное заболевание, то в его основании лежит не мутация определенного гена, а дефект всей хромосомы, то есть, целой молекулы ДНК, а именно — дополнительная хромосома.
Вероятность развития хромосомных патологий напрямую зависит от возраста родителей, что доказано в ходе крупных научных исследований. Особенно важное значение имеет возраст матери — после 40 лет возможность зачать больного ребенка увеличивается примерно в 7 раз в сравнении с общей популяцией. Возраст отца также важен, но здесь зависимость выражена не так сильно.

Признаки и симптомы синдрома Эдвардса
Симптомы синдрома Эдвардса во многом зависят от формы заболевания у конкретного больного.
На сегодняшний день генетики различают три типа болезни:
Дети в случае наличия такого изменения количества хромосом рождаются со сниженной массой тела и наличием множественных пороков развития, которые заключаются в изменении формы черепа, уменьшении диаметра рта, аномалии твердого неба, наличии узких глазных щелей, деформации ушных раковин, дефекты конечностей, половых органов, специфические дерматоглифические проявления. Также часто развиваются пороки сердца, которые заключаются в нарушении заращения перегородок и формирования клапанов между его полостями.
Благодаря яркой клинической картине диагностика заболевания не представляет серьезных трудностей:
В зависимости от степени выраженности различают 4 варианта порока:
Деформация стопы в виде стопы-качалки обусловлена неправильным расположением костей. При этом пятка сильно выступает назад, свод стопы отсутствует. Вогнутая линия, которая в норме соединяет пятку и большой палец, отсутствует, нередко стопа выпуклая, из-за чего похожа на ножки кресла-качалки, что и обусловило название деформации.
При наличии стопы-качалки часто наблюдается непропорциональное строение пальцев ног. Большой палец у больных детей короткий — короче, чем второй палец стопы. Важно понимать, что у взрослых этот признак не имеет диагностической ценности, так как может появиться из-за других заболеваний, неправильной обуви, суставной аномалии. Вместе с непропорциональным строением нередко имеет место сращение пальцев. В легких случаях это лишь кожная складка, но нередко обнаруживают и костные дефекты.
Кисти малыша находятся в специфическом положении — они сжаты в кулак, но мизинец и указательный палец при этом располагаются поверх среднего и безымянного. Это так называемое флексорное положение кисти, обусловленное повышенным тонусом мышц-сгибателей.
Недоразвитая нижняя челюсть встречается у 70% детей, которым поставлен диагноз «синдром Эдвардса». Подбородок сильно втянут, что заметно невооруженным глазом. Ребенок не может держать рот закрытым, что ведет к вытеканию слюны. У выживших детей формируется неправильный прикус.
Из аномалий половых органов чаще всего имеют место недоразвитие полового члена и увеличение клитора. Также возможно неправильное расположение уретры. У мальчиков яички могут оставаться в брюшной полости или паховом канале. В большинстве случаев эти отклонения сочетаются с пороками внутренних половых органов и мочевого пузыря.
Дерматоглифические симптомы — характерные отклонения в расположении узоров и складок на ладонях. В большинстве случаев дерматоглифика используется как предварительная диагностика синдрома Эдвардса, особенно частичной и мозаичной форм. При полной трисомии клиническая картина выражена настолько ярко, что потребность в детальном изучении узора ладоней отпадает за ненадобностью.
Важно понимать, что сами по себе вышеперечисленные признаки — далеко не диагноз. Единичные отклонения могут быть вариантом нормы у абсолютно здоровых людей, но наличие нескольких наиболее часто встречающихся отклонений является поводом для более детального обследования.
Диагностика синдрома Эдвардса
Диагностика синдрома Эдвардса заключается в генетическом исследовании, которое может проводиться во время беременности. При этом выполняется цитологическое исследование околоплодных вод с определением количества хромосом клеток эпидермиса плода. Так как подобные манипуляции провоцируют определенные осложнения, синдром Эдвардса с их помощью диагностируют только пациентам из группы риска.

Лечение синдрома Эдвардса
Профилактика синдрома Эдвардса
Профилактика синдрома Эдвардса сводится к тщательному планированию предстоящей беременности. Всем парам из группы риска настоятельно рекомендуют проконсультироваться у генетика. Врач детально изучит семейный анамнез пары, выявит факторы риска, проведет генетический анализ пары. При сборе семейного анамнеза доктор расспросит о наследственных заболеваниях в семье. При наличии таковых, вероятность рождения ребенка с аномалией возрастает до 1%. Очень желательно, чтобы родители собрали максимум информации о своих предках. Затем генетик выявит факторы риска и даст рекомендации по их устранению, если это возможно.
Генетический анализ необходим для обнаружения дефектных генов в молекуле ДНК. Чем больше мутаций будет выявлено, тем выше вероятность рождения больного ребенка. Всем беременным из группы риска показана пренатальная диагностика, благодаря которой можно вовремя диагностировать болезнь и принять решение по поводу дальнейшего вынашивания ребенка. В принципе, это также профилактика синдрома Эдвардса.
Из-за множественных пороков развития примерно половина детей умирает в первые три месяца жизни. Только 10 детей из 100 доживут до года. Более старшие дети нуждаются в тщательном уходе, постоянном лечении и профилактике осложнений синдрома Эдвардса. При мозаичной или частичной форме прогноз более благоприятный. Длительность жизни детей больше, но они также нуждаются в уходе. При полноценных реабилитационных мероприятиях малыш может научиться сидеть и питаться, иногда хорошо выражена реакция на окружающих. Из-за серьезной умственной отсталости больные не способны устроиться даже на простейшую работу.
Синдром Эдвардса можно предотвратить, если вы беременеете с помощью ЭКО, используя диагностическую процедуру под названием ПГД (преимплантационная генетическая диагностика). Она дает возможность выявить хромосомную аномалию еще до помещения эмбриона в полость матки, то есть, до наступления беременности. Для переноса будет выбран тот эмбрион, который гарантированно не имеет никаких генных или хромосомных мутаций.
Заключение
Теоретически риск появления ребёнка с синдромом Эдвардса есть в любой супружеской паре. Также известно, что эта вероятность намного выше у возрастных родителей. Именно поэтому с целью своевременного обнаружения хромосомной аномалии у плода не стоит пренебрегать антенатальным скринингом, который входит в программу ведения беременности.
У вас есть вопросы? Проконсультируйтесь с нашими опытными врачами и эмбриологами.
При изучении хромосом метафазной пластинки можно диагностировать болезнь эдвардса
НИПТ – это скрининг-тест ДНК, который поможет узнать важную информацию о вашей беременности.
С помощью этого теста, вы сможете:


Преимущества НИПТ:


В отличие от остальных скринингов при беременности, тест Panorama дает возможность:
Некоторые методы генетического тестирования, такие как амниоцентез, кордоцентез или биопсия хориона (CVS), являются инвазивными (в частности, осуществляется прокол передней брюшной стенки беременной тонкой пункционной иглой) и поэтому несут риск выкидыша. Альтернативой ему служит пренатальный тест Panorama, который требует только забора венозной кровии совершенно безопасен для матери и ребенка.
Вы получаете полную и точную информацию о протекании Вашей беременности без риска!
Трисомия хромосомы 18 (Синдром Эдвардса)
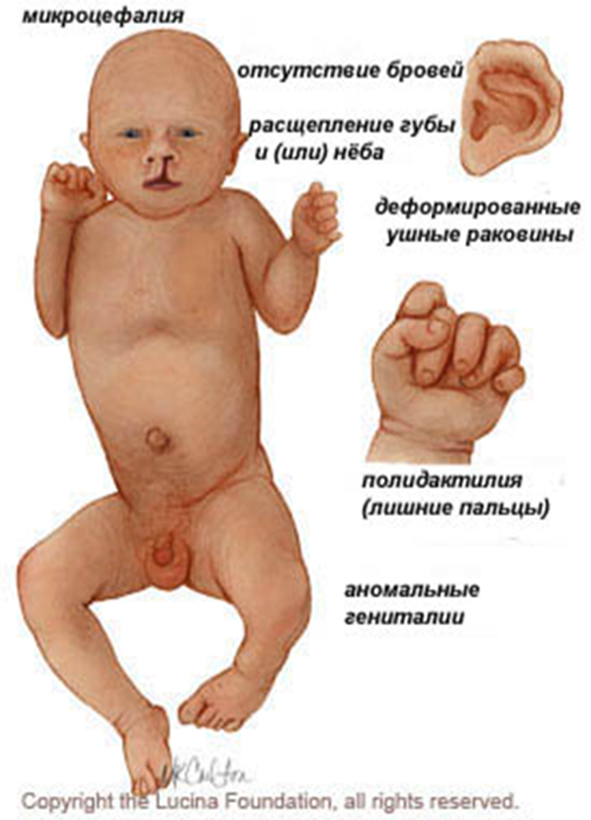
Клинические особенности, связанные с трисомии 18 включают, но не ограничиваются следующими: расстройства центральной нервной системы (голопрозенцефалия, менингомиелоцеле), пороки развития глаз, носа, расщелина губы и / или неба, деформация ушных раковин, деформированные конечностей, полидактилия, а также дефекты сердца, половых органов, и пр.
ЭТИОЛОГИЯ
Причиной истинной трисомии 18 является нерасхождение хромосом при формировании яйцеклетки и сперматозоида, когда одна гамета получает дополнительную 18 хромосому. Нерасхождение может произойти в первом (MI) или втором (MII) мейотического этапе.
В 90-97% случаев дополнительная хромосома 18 имеет материнское происхождение, а отцовское происхождение в 3-10% всех случаев. Среди случаев трисомии 18 материнского происхождения, 31-39% бывают в результате нерасхождения хромосом в фазе MI и 61-69% в результате нерасхождение в фазе MII (Bugge et al., 1998; Nicolaidis and Petersen, 1998; Eggermann et al., 1996; Ramesh and Verma, 1996; Fisher et al., 1995; Jacobs and Hassold, 1995; Fisher et al., 1993; Ya-gang et al., 1993).
ДЕМОГРАФИЯ И РЕПРОДУКТИВНОСТЬ
Риск трисомии 18, как известно, возрастает с увеличением возраста матери (Munne 2004, Naguib 1999, Baty 1994, Buyse 1990, Goldstein 1988, Schreinemachers 1982). Риск трисомии 18 (синдрома Эдвардса) связывают также и с увеличением отцовского возраста, однако, если риск трисомии 18 увеличивается за счет возраста матери, то возрастом отца в таких случаях пренебрегают. (Naguib 1999, Baty 1994). Для женщин старше 45 лет риск родить больного ребёнка составляет 0,7 %.
Раса/этническая принадлежность практически не влияет на риск трисомии 18 (Buyse 1990). Одно исследование показало, что из четырех расовых/этнических групп обследованных (европейцы, дальневосточные азиаты, жители островов тихого океана, филиппинцы), риск трисомии 18 был самым высоким для дальневосточных азиатов и самым низким для жителей островов тихого океана (Forrester, 1999). Тем не менее, эти различия в риске по видимому, связаны с различиями в распределении материнского возраста среди этих расовых / этнических групп.
Географическая область может влиять на риск трисомии 18. В одном исследовании приводятся данные, что риск трисомии 18 выше среди городских жителей (Forrester, 1999). Этот повышенный риск остается после учета возраста матери. Несколько исследований показали, что на распространенность трисомии 18 могут влиять сезонные колебания (Naguib, 1999).
Некоторые исследования показали, что Трисомия 18 (синдром Эдвардса) в последние годы имеет тенденцию к увеличению. Однако, возможно, это связано как с улучшением диагностики анеуплоидий, в т.ч. пренатальной диагностики и диагностики фетусов (Pradat 1991), так и с повышением возраста рожающих женщин.(Gessner 2003, Forrester, 1999).
За последние несколько десятилетий, было установлено, что в сыворотке женщин с плодом с трисомией 18 имеется низкий уровень альфа-фетопротеина, хорионический гонадотропин человека, и эстриол (anick 1993, Greenberg 1992, Doran 1986). Кроме того, при пренатальном ультразвуковом исследовании обнаруживаются различные структурные аномалии часто связаны с трисомией 18 (Abramsky 1993, Vintzileos 1987). Пренатальный скрининг маркеров, УЗИ и окончательный диагноз подтвержденный кариотипированием с помощью таких процедур, как амниоцентез и биопсия хориона, позволяют определить трисомию 18 в период внутриутробного развития.
Пол ребенка влияет на риск трисомии 18. Девочки с трисомией 18 рождаются в 3 раза чаще мальчиков. (Forrester 1999, Naguib 1999, Carothers 1999, Huether 1996, Pradat 1991, Buyse 1990, Goldstein 1988). Одно исследование показало, что соотношение полов изменяется в различных расовых /этнических группах, однако это такие наблюдения сделаны на небольших выборках, что не позволяет говорить о такой связи достоверно.(Huether 1996).
Риск повторения для трисомии 18 составляет, примерно, 1% (Baty 1994, Buyse 1990). Последние исследования показали, что риск трисомии увеличивается у женщин, которые имели трисомии 18 в предыдущих беременностях, независимо от рождения живого ребенка или внутриутробной гибели плода. То есть, даже если беременность самопроизвольно прерывается, риск остается повышенным (Munne 2004). Существует также повышенный риск трисомной беременности у женщин, с низким числом овоцитов (Kline, 2000). Вероятно, это условие связано с наступлением менопаузы.
ОБРАЗ ЖИЗНИ И ФАКТОРЫ ОКРУЖАЮЩЕЙ СРЕДЫ.
Вероятность хромосомных ошибок в настоящее время может увеличиваться из-за вспомогательных репродуктивных технологий (ВРТ). Однако, до конца не ясно, связано ли это с несовершенством лабораторных методов или с генетическими нарушениями у родителей, которые одновременно могут являться и причиной бесплодия. То есть пары, которые не могут забеременеть естественным путем могут быть предрасположены к генетическим ошибкам (Palermo 2000).
Одно исследование показало, что различия в метаболизме фолиевой кислоты у женщин не связаны с мейотическими ошибками приводящими к анеуплоидиям.
Регулярное употребление поливитаминов не связано с уменьшением риска трисомии 18 (Botto 2004).
ГЕНЕТИЧЕСКАЯ ПРЕДРАСПОЛОЖЕННОСТЬ
Иногда, вследствие явления, которое называется сбалансированная транслокация, некоторые люди могут нести генетический материал, принадлежащий хромосоме 18 в другой хромосоме. Поскольку у таких людей нет дополнительного генетического материала они не имеют признаков трисомии 18. Однако, у таких людей есть повышенный риск рождения детей с этим заболеванием. Установить носительство сбалансированной хромосомной транслокации можно при исследовании кариотипа.
В редких случаях один из родителей может быть носителем частичной трисомии 18, которая может передаваться по наследству. Установить носительство частичной трисомии можно с помощью хромосомного микроматричного анализа.
Таким образом, в большинстве случаев синдром Эдвардса является результатом случайных ошибок в делении клетки и мало связан с какими либо факторами окружающей среды или состояния человека.



