Увеличение auc что это
AUC — аббревиатура от англоязычного Area Under the Curve (площадь под кривой). В медицинской и фармацевтической обычно используется без перевода.
AUC в фармакокинетике
AUC — фармакокинетический параметр, характеризующий суммарную концентрацию лекарственного препарата в плазме крови в течение всего времени наблюдения. Математически определяется как интеграл от 0 до ∞ функции концентрации препарата (фармакокинетической кривой) в плазме крови от времени и равен площади фигуры, ограниченной фармакокинетической кривой и осями координат. Параметр AUC связан с другими фармакокинетическими параметрами — объемом распределения, общим клиренсом. При линейности кинетики препарата в организме величина AUC пропорциональна общему количеству (дозе) препарата, попавшего в системный кровоток.

AUC T — площадь под частью фармакокинетической кривой, от начала исследования (t = 0) до некоторого времени t = T (обычно заданное в часах). Например, AUC24 равен площади под фармакокинетической кривой в течение первых 24-х часов исследования.
AUC в исследовании кислотности органов желудочно-кишечного тракта
AUC [H + ] или просто AUC («интегральная кислотность») — широко используемое в зарубежных работах обозначение показателя, применяемого для оценки кислотоподавляющих свойств лекарственных препаратов и равного площади под заданным участком рН-метрической кривой. В отечественной литературе этот показатель называется «площадь ощелачивания» (Рапопорт С.И. и др.). AUC [H + ] равен S (iн, iк, 0) работы «Математический анализ компьютерных рН-грамм верхних отделов ЖКТ (п. 4 на стр. 2)».
Увеличение auc что это
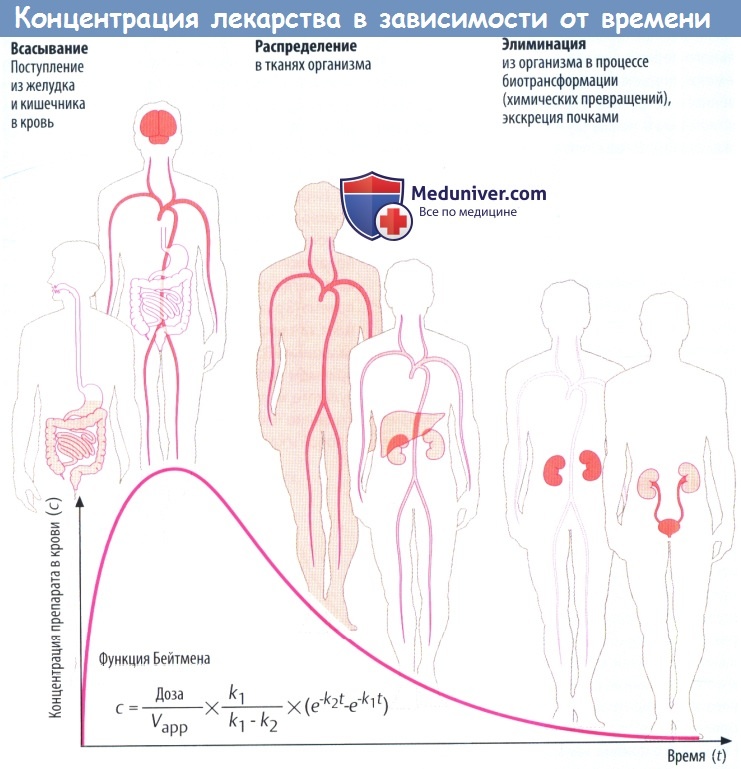
(А) Лекарственные средства попадают в организм и выводятся из него разными путями. Таким образом, организм представляет собой открытую систему, в которой фактическая концентрация препарата отражает взаимодействие между его поступлением (приемом) и эвакуацией (элиминацией).
Скорость всасывания препарата в желудке и кишечнике зависит от множества факторов: скорости растворения вещества (в случае приема твердой лекарственной формы) и транзита по ЖКТ, проницаемости слизистой для препарата, его градиента концентрации на границе слизистой и крови,кровоснабжения слизистой оболочки.
Всасывание из кишечника приводит к повышению концентрации лекарственного вещества в крови. Препарат разносится с кровью к различным органам (распределение), которые поглощают его в количестве, соответствующем его химическим свойствам и скорости кровотока через орган.
Например, органы с хорошим кровоснабжением, такие как головной мозг, получают большее количество препарата, чем органы с низким кровоснабжением. В результате поглощения тканями происходит снижение концентрации лекарственного вещества в крови. По мере снижения градиента на границе слизистой оболочки и крови всасывание в кишечнике замедляется. Пик концентрации в крови достигается тогда, когда количество вещества, покидающего кровь за единицу времени, равно количеству всосавшегося.
Поступление вещества в ткани печени и почек представляет собой перемещение в органы выведения. Концентрация препарата в крови в различные периоды времени представляет собой совокупность процессов абсорбции, распределения и элиминации, которые пересекаются во времени.
Если распределение происходит значительно быстрее, чем элиминация, снижение концентрации в крови вначале происходит быстро, а затем замедляется. Фаза быстрого снижения обозначается как α-фаза (фаза распределения), медленного — как β-фаза (фаза элиминации). Если препарат распределяется быстрее, чем абсорбируется, концентрацию препарата в крови можно описать математически упрощенной функцией Бейтмена (k1 и k2 — константы скорости для абсорбции и элиминации соответственно).
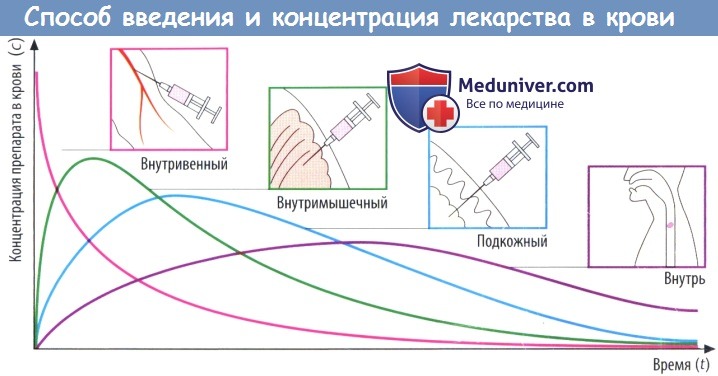
(В) Скорость абсорбции зависит от способа введения препарата. Чем выше скорость абсорбции, тем короче будет время (tmax), которое требуется для достижения пика концентрации в плазме (cmax), тем выше будет cmax и тем раньше уровень препарата в крови снова начнет снижаться.
Площадь под кривой, описывающей зависимость концентрации препарата в крови от времени (AUC), не зависит от пути введения препарата при условии, что доза и биодоступность остаются теми же (закон соответственных состояний). Таким образом, AUC можно использовать для вычисления биодоступности (F) препарата.
Значение AUC, измеренное после приема внутрь и в/в введения определенной дозы конкретного лекарственного вещества, соответствует проценту вещества, попавшего в системный кровоток после приема внутрь: F = AUCприем внутрь/AUCв/в введение.
Определение концентрации препарата в крови позволяет сравнить различные патентованные лекарственные средства, содержащие одно и то же действующее вещество в одинаковой дозе. Идентичные кривые зависимости концентрации в крови от времени для препаратов различных производителей (при условии стандартных лекарственных форм) означают биоэквивалентность стандартного вещества и нового исследуемого препарата.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Глубокое погружение в ROC-AUC
Я думаю, что большинство людей слышали о ROC-кривой или о AUC (площади под кривой) раньше. Особенно те, кто интересуется наукой о данных. Однако, что такое ROC-кривая и почему площадь под этой кривой является хорошей метрикой для оценки модели классификации?
Теория ROC-кривой
Полное название ROC — Receiver Operating Characteristic (рабочая характеристика приёмника). Впервые она была создана для использования радиолокационного обнаружения сигналов во время Второй мировой войны. США использовали ROC для повышения точности обнаружения японских самолетов с помощью радара. Поэтому ее называют рабочей характеристикой приемника.
AUC или area under curve — это просто площадь под кривой ROC. Прежде чем мы перейдем к тому, что такое ROC-кривая, нужно вспомнить, что такое матрица ошибок.
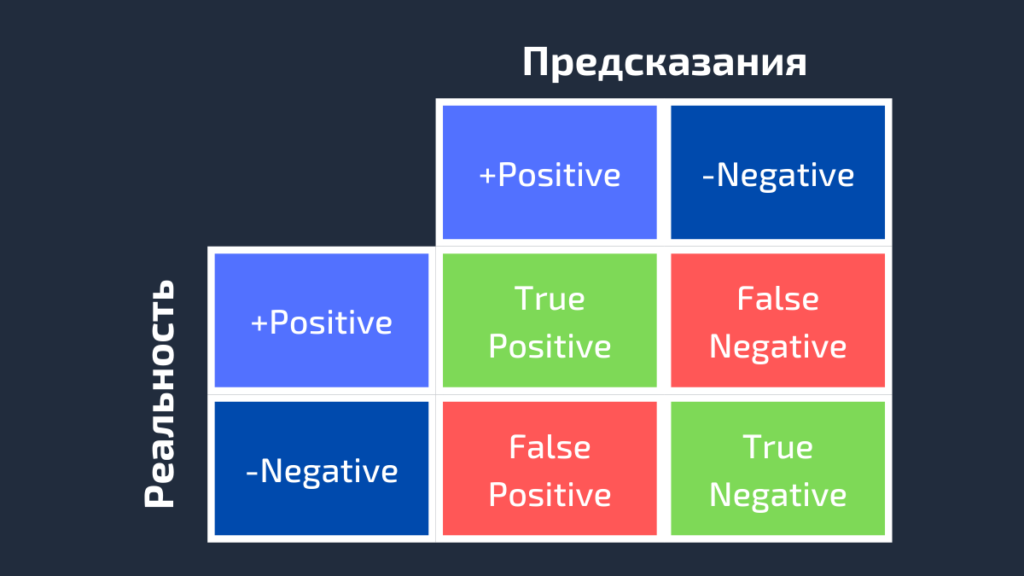
Как видно из рисунка выше, матрица ошибок — это комбинация вашего прогноза (1 или 0) и фактического значения (1 или 0). В зависимости от результата предсказания и того, корректна ли была проведена классификация, матрица разделена на 4 части. Например, true positive (истинно положительный) результат — это количество случаев, в которых вы правильно классифицируете семпл как положительный. А false positive (ложноположительный) — это число случаев, в которых вы ошибочно классифицируете семпл как положительный.
Матрица ошибок содержит только абсолютные числа. Однако, используя их, мы можем получить множество других метрик, основанных на процентных соотношениях. True Positive Rate (TPR) и False Positive Rate (FPR) — две из них.
True Positive Rate (TPR) показывает, какой процент среди всех positive верно предсказан моделью.
TPR = TP / (TP + FN).
False Positive Rate (FPR): какой процент среди всех negative неверно предсказан моделью.
FPR = FP / (FP + TN).
Хорошо, давайте теперь перейдем к кривой ROC!
Что такое ROC-кривая?
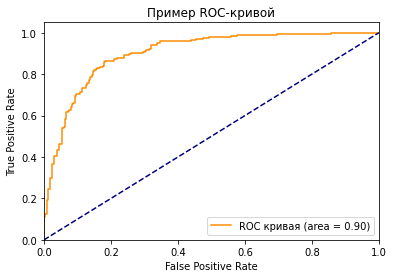
Как вы можете видеть на графике, кривая ROC — это просто отношение TPR к FPR. Теперь вам все понятно, в заключение…
Поверили?
Если серьезно, вы можете прочитать намного больше информации из диаграммы. Первый вопрос, который я хочу здесь обсудить: у нас же есть только один набор TPR, FPR, посчитанный на основе сделанных моделью предсказаний. Так откуда взялось такое количество точек для построения целого графика?
Все следует из того, как работает модель классификации. Когда вы строите классификационную модель, такую как дерево решений, и хотите определить, будут ли акции расти в цене или падать на основе входных данных. Модель сначала рассчитает вероятность увеличения или уменьшения, используя предоставленные вами исторические данные. После этого, основываясь на пороговом значении, она решит, будет ли результат увеличиваться или уменьшаться.
Да, ключевое слово здесь — порог. Разные пороговые значения создают разные TPR и FPR. Они представляют те самые точки, что образуют кривую ROC. Вы можете выбрать «Увеличение» в качестве предсказания модели, если полученная на основе исторических данных вероятность роста акций больше 50%. Также можете изменить пороговое значение и отобразить «Увеличение», только если соответствующая вероятность больше 90%. Если вы установите 90% порог вместо 50%, вы будете более уверены в том, что выбранные для «Увеличения» акции действительно вырастут. Но так вы можете упустить некоторые потенциально выгодные варианты.
Что значит синяя пунктирная линия на графике?
Как мы знаем, чем больше площадь под кривой (AUC), тем лучше классификация. Идеальная или наилучшая кривая — это вертикальная линия от (0,0) до (0,1), которая тянется до (1,1). Это означает: модель всегда может различить положительные и отрицательные случаи. Однако, если вы выбираете класс случайным образом для каждого семпла, TPR и FPR должны увеличиваться с одинаковой скоростью. Синяя пунктирная линия показывает кривую TPR и FPR при случайном определении positive или negative для каждого случая. Для этой диагональной линии площадь под кривой (AUC) составляет 0.5.
Что произойдет с TPR, FPR и ROC-кривой, если изменить пороговое значение?
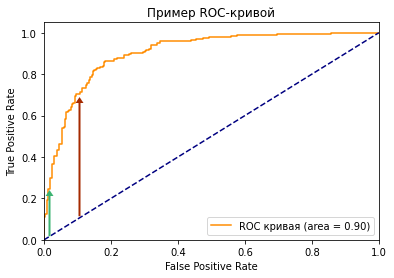
Посмотрите на две точки на ROC-кривой. Зеленая точка имеет очень высокий порог, это означает, что только если вы уверены на 99%, можете классифицировать случай как positive. Красная точка имеет относительно более низкий порог. Это означает, что вы можете классифицировать случай как positive, если вы уверены на 90%.
Как изменяются TPR и FPR при движении от зеленой точки к красной?
И TPR, и FPR увеличиваются. Когда вы уменьшаете порог, модель будет определять больше положительных случаев. Таким образом, TP увеличивается, как и TP/(TP + FN). С другой стороны, вы неизбежно ошибочно классифицируете некоторые отрицательные случаи как положительные из-за снижения порога, и поэтому FP и FP/(FP + TN) также увеличиваются.
Мы видим, что TPR и FPR положительно коррелируют. Вам нужно балансировать между максимальным охватом positive случаев и минимизацией неправильной классификации negative случаев.
Как выбрать оптимальную точку на кривой ROC?
Трудно определить оптимальную точку, потому что нужно выбрать наиболее подходящее пороговое значение, учитывая сферу применения модели. Однако общее правило — максимизировать разницу (TPR-FPR), которая на графике представлена вертикальным расстоянием между оранжевой и синей пунктирной линией.
Почему площадь под кривой ROC – хорошая метрика для оценки модели классификации?
Хорошая метрика модели машинного обучения должна отображать истинную и постоянную способность модели к прогнозированию. Это означает, что, если я изменю тестовый набор данных, он не должен давать другой результат.
ROC-кривая учитывает не только результаты классификации, но и вероятность предсказания всех классов. Например, если результат корректно классифицирован на основе 51% вероятности, то он, скорее всего, будет классифицирован неверно, если вы воспользуетесь другим тестовым датасетом. Кроме того, ROC-кривая также учитывает эффективность модели при различных пороговых значениях. Она является комплексной метрикой для оценки того, насколько хорошо разделяются случаи в разных группах.
Какое значение AUC является приемлемым для модели классификации?
Как я показал ранее, для задачи двоичной классификации при определении классов случайным образом, вы можете получить 0.5 AUC. Следовательно, если вы решаете задачу бинарной классификации, разумное значение AUC должно быть > 0.5. У хорошей модели классификации показатель AUC > 0.9, но это значение сильно зависит от сферы ее применения.
Как рассчитать AUC и построить ROC-кривую в Python?
Если вы просто хотите рассчитать AUC, вы можете воспользоваться пакетом metrics библиотеки sklearn (ссылка).
Если вы хотите построить ROC-кривую для результатов вашей модели, вам стоит перейти сюда.
Вот код для построения графика ROC, который я использовал в этой статье.
Изучение биоэквивалентности лекарственных средств как одного из видов клинических испытаний
В настоящее время в Украине значительно возросло количество заявок на регистрацию отечественных и зарубежных лекарственных препаратов. Подавляющее большинство из них — генерические препараты, т.е. лекарственные средства, выпускаемые различными фармацевтическими компаниями после прекращения срока действия патента на оригинальный препарат.
Клиническая практика показала, что препараты, имеющие одни и те же активные вещества и даже лекарственные формы и дозы, но выпускаемые различными производителями, существенно различаются как по терапевтической эффективности, так и по частоте и выраженности вызываемых ими побочных эффектов (Белоусов Ю.Б., Моисеев В.С., Лепахин В.К., 1997).
В последние годы в результате развития фармакологии, внедрения высокочувствительных методов исследования, таких, как жидкостная и газовая хроматография, радиоиммунный и ферментно-химический анализ, стало возможным в полной мере понять и оценить роль особенностей технологии производства, качественного и количественного состава вспомогательных веществ лекарственной формы и многих других факторов в действии лекарственных средств.
В большинстве случаев различия в терапевтической эффективности препаратов, содержащих одни и те же активные вещества, обусловлены изменением их биодоступности — количества лекарственного вещества, которое попадает в системный кровоток, и скорости, с которой этот процесс происходит. В связи с этим возникло новое понятие — биоэквивалентность.
Два лекарственных препарата считают биоэквивалентными, если они фармацевтически эквивалентны, имеют одинаковую биодоступность и после назначения в одинаковой дозе являются сходными, обеспечивая должную эффективность и безопасность (WHO, 1994, 1996).
Оценку биоэквивалентности лекарственных средств в настоящее время считают одним из основных методов медико-биологического контроля качества генерических препаратов, т.е. таких лекарственных средств, которые содержат одно и то же активное вещество в одинаковой дозе и в той же лекарственной форме, что и соответствующее оригинальное средство.
Фармакотерапевтическое действие препарата зависит от многих факторов (биодоступность, состояние печени и почек и т.п.). Особенно трудно бывает предсказать возможную концентрацию лекарственного средства в крови при:
· наличии сопутствующих заболеваний;
· применении взаимодействующих препаратов;
· нарушении всасывания и низкой биодоступности;
· заболевании печени и почек;
· нарушении связывания лекарственных веществ с белками крови;
· генетически обусловленных особенностях метаболизма препаратов.
В таких ситуациях концентрация препарата может оказаться слишком низкой или слишком высокой. В первом случае снижается эффективность лечения, во втором — повышается риск возникновения побочных реакций.
В связи с этим при исследовании биоэквивалентности необходимо очень тщательно подходить к подбору его участников.
Прежде всего необходимо подчеркнуть, что изучение биоэквивалентности — это клинические испытания, где субъектом исследования является человек. Поэтому к таким исследованиям предъявляют те же требования, что и ко всем другим клиническим испытаниям.
Изучение биоэквивалентности следует проводить в соответствии с принципами Надлежащей клинической практики в целях гарантии качества представляемых данных и защиты прав, здоровья и благополучия исследуемых.
ВОЗ рекомендует следующие подходы к отбору испытуемых (1994).
Контингент исследуемых для изучения биоэквивалентности должен быть максимально однородным. Чтобы снизить разброс получаемых данных, в основном это должны быть здоровые добровольцы. Должны быть представлены точные критерии включения/исключения. Желательно, чтобы в исследовании участвовали мужчины и женщины (для женщин оценку риска нужно определять индивидуально и они должны быть предупреждены о возможном риске для плода в случае беременности). В исследование включают лиц в возрасте от 18 до 55 лет. Масса их тела должна быть в пределах возрастной нормы для данного пола. Предпочтительно, чтобы испытуемые были некурящими. В противном случае они должны быть идентифицированы как таковые. Пригодность добровольцев к участию в исследовании должна быть подтверждена с помощью стандартных лабораторных тестов, данных анамнеза и физического обследования. До и в процессе исследования можно проводить специальные медицинские обследования, необходимость которых обусловлена особенностями фармакологических свойств изучаемого препарата.
В некоторых случаях корректнее включать в исследование пациентов, а не здоровых добровольцев. Такая ситуация может возникнуть, если исследуемое лекарственное средство обладает известными побочными действиями и здоровью добровольцев может быть нанесен серьезный ущерб (например, при изучении лекарственных препаратов, используемых в онкологии, при лечении ВИЧ-инфекции).
При исследовании биоэквивалентности минимальное число испытуемых — 12 человек.
Проведение исследования и его дизайн должны базироваться на знаниях фармакокинетики и фармакодинамики исследуемого лекарственного вещества.
При этом должны быть обеспечены стандартные условия для испытуемых на период проведения исследования, а именно:
· пищевой и водный режим (стандартная диета);
· полное исключение или ограничение приема лекарственных средств до и в период проведения исследования;
· исключение употребления алкоголя, кофеина, наркотических средств, концентрированных соков;
· время пребывания в исследовательском центре;
· время окончания исследования.
Особенностью дизайна таких исследований является то, что каждый испытуемый получает как стандартный препарат, так и тестируемый. Отдают предпочтение перекрестному методу с рандомизированным распределением добровольцев.
При изучении биоэквивалентности лекарственных препаратов наиболее важными являются следующие фармакокинетические параметры (рис.1):
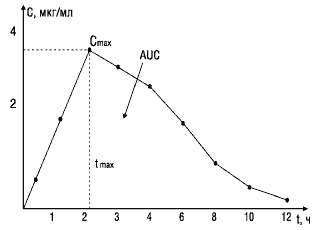
Рис.1. Основные параметры фармакокинетики, используемые при изучении биоэквивалентности лекарственных веществ
Cmax — максимум, или пик концентрации лекарственного вещества в крови;
tmax — время достижения максимальной концентрации вещества в плазме крови;
AUC — площадь под фармакокинетической кривой — кривой «концентрация—время» (изменение концентрации активного вещества в плазме или сыворотке крови во времени).
Значение показателя максимальной концентрации вещества можно объяснить с помощью следующего примера. На рис. 2 представлены фармакокинетические кривые двух лекарственных препаратов. Кривая 1 характеризует концентрацию в крови стандартного препарата, кривая 2 — тестируемого. Горизонтальной линией отмечена минимальная эффективная концентрация, при которой данное вещество оказывает терапевтическое действие. Как видно, Сmax тестируемого препарата (кривая 2) не достигает уровня минимальной эффективной концентрации и, следовательно, не оказывает терапевтического действия.
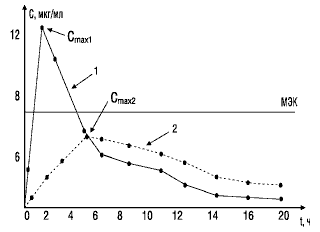
Рис. 2. Сопоставление максимальных концентраций в крови двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; Сmax1, Сmax2 — соответствующие максимальные концентрации сравниваемых препаратов в крови
Второй важный параметр — время достижения максимальной концентрации лекарственного вещества в крови. Этот показатель отражает скорость его всасывания и скорость наступления терапевтического эффекта. На рис. 3 показано, что Сmax стандартного препарата (кривая 1) достигается через 1 ч, а тестируемого (кривая 2) — через 4 ч. Такое различие во времени достижения максимальной концентрации препарата в крови может обусловить изменение клинических показаний к применению данного препарата.
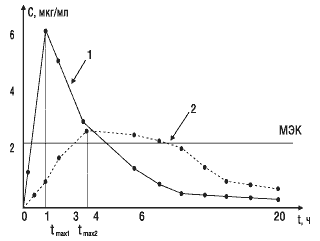
Рис.3. Сопоставление времени достижения максимальной концентрации двух препаратов: МЭК — минимальная эффективная концентрация; 1 — стандартный препарат; 2 — тестируемый препарат; tmax1, tmax2 — соответствующее время достижения максимальных концентраций сравниваемых препаратов в крови
Третьим важным параметром биоэквивалентности является площадь под фармакокинетической кривой, которая отражает количество вещества, поступившего в кровь после однократного введения препарата. На рис. 3 показано, что две кривые имеют разную форму, разные пики и неодинаковое время достижения максимальной концентрации. Но площади под этими кривыми близки по величине, следовательно, оба препарата обеспечивают поступление в кровь одинакового количества лекарственного вещества.
На рис. 4 представлен другой пример соотношения кривых, отражающих кинетику двух сравниваемых препаратов. Площадь под кривой 1 практически в два раза больше, чем под кривой 2. Обращает на себя внимание то, что максимальная концентрация и время ее достижения схожи у стандартного и тестируемого препаратов. Однако площадь под фармакокинетической кривой у тестируемого препарата в 2 раза меньше за счет более быстрого выведения его из крови. В данном случае можно ожидать уменьшения длительности действия лекарственного препарата и снижения его терапевтического эффекта.
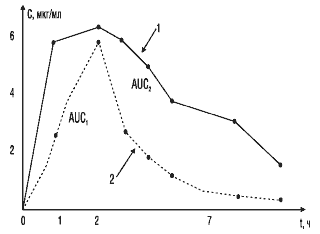
Рис. 4. Сопоставление площадей под фармакокинетическими кривыми двух препаратов: 1 — стандартный препарат; 2 — тестируемый препарат; AUC1, AUC2 — соответствующие площади под кривыми «концентрация—время» сравниваемых препаратов
Таким образом, два препарата считают биоэквивалентными, если они имеют схожие фармакокинетические показатели. По регламенту ВОЗ (1994, 1996) и ЕС (1992) их различие не должно превышать 20%.
Необходимо отметить, что планировать и проводить исследование по определению биоэквивалентности должен коллектив специалистов различных профилей: клинические фармакологи, врачи-клиницисты, биохимики, химики-аналитики, биостатистики. Все этапы проведения исследования должны быть тщательно описаны, проанализированы и представлены в подробном отчете.
Внедрение определения биоэквивалентности как метода позволяет сделать обоснованное заключение о качестве, эффективности и безопасности сравниваемых препаратов на основании меньшего объема первичной информации и в более сжатые сроки, чем при проведении клинических испытаний.
В настоящее время уже существуют регламенты изучения биоэквивалентности ВОЗ (1996), ЕС (1992), Российской Федерации (1995). В них изложены основные обоснования необходимости проведения таких исследований.
Так, изучение биоэквивалентности проводят, если:
· существует риск отсутствия биоэквивалентности и/или
· существует риск снижения фармакотерапевтического действия и клинической безопасности препарата.
Например, обязательно оценивают:
· препараты для лечения состояний, при которых необходим гарантированный терапевтический эффект;
· препараты с узким терапевтическим коридором безопасности;
· препараты, фармакокинетика которых осложнена снижением абсорбции менее 70% или с высокой элиминацией (более 70%);
· препараты с неудовлетворительными физико-химическими свойствами (низкая растворимость, нестабильность, полиморфизм);
· препараты с документированно подтвержденным существованием проблем биодоступности.
В настоящее время в Украине достаточно широко используют высокоэффективные методы для определения фармакокинетических параметров. Расширение имеющейся материально-технической базы, разработка регламента проведения исследований биоэквивалентности лекарственных веществ, подготовка специалистов в этой области позволят решить актуальную задачу по оценке эффективности и безопасности генерических препаратов отечественного и зарубежного производства.
В.И. Мальцев, А.П. Викторов,
В.Н. Коваленко, Т.К. Ефимцева, Л.И. Ковтун
Государственный фармакологический центр Министерства здравоохранения Украины, Киев
ЛИТЕРАТУРА
Белоусов Ю.Б., Моисеев В.С., Лепахин В.К. (1997) Клиническая фармакология и фармакотерапия. Универсум Паблишинг, Москва, 530 с.
Страчунский Л.С., Фирсов А.А., Белоусов Ю.Б., Рудаков А.Г., Судиловский С.Д. (1995) Правила проведения исследований биоэквивалентности лекарственных средств. Часть 1. Организационные аспекты испытаний на здоровых добровольцах. Москва, 16 с.
Фирсов А.А., Родионов А.П., Страчунский Л.С., Рудаков А.Г., Барманова Е.Ю. (1995) Правила проведения исследований биоэквивалентности лекарственных средств. Часть 1. Фармакокинетические аспекты. Москва, 11 с.
Холодов Л.Е., Яковлев В.П. (1985) Клиническая фармакокинетика. Медицина, Москва, 464 с.
Consultative Document. Interchangeable multy-source pharmaceutical products: WHO draft guideline on marketing authorization requirements (1994) WHO Drug Information, 8(2): 71–83.
Investigation of bioavailability and bioequivalence (1992) The rules governing medicinal products in the European Community, III: 149–168.
Multisource (generic) pharmaceutical products: guidelines on registration requuirements to establish interchangeability (1996) WHO Technical Report Series, 863: 114–154.



