Фармакокинетика
Содержание
Фармакокинетика [ править | править код ]
Предметом фармакокинетики является анализ всех факторов, определяющих абсорбцию, распределение, метаболизм и экскрецию лекарственных веществ. Термин отражает скорость перемещения (кинетика) лекарств (фармако) в организме и вне его.
Однокомпартментная модель [ править | править код ]
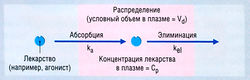
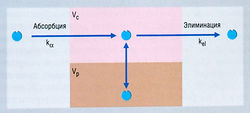
Согласно простейшей модели, организм рассматривается как единое однородное пространство (компартмент), в которое лекарство вводят и из которого оно элиминируется (рис. 4.10). Предполагается, что введенное лекарство сразу же распределяется по всему пространству. Если элиминация является кинетическим процессом первого порядка, то ее скорость пропорциональна концентрации в плазме, т.е. лекарство выводится экспоненциально. Математическое уравнение для этого экспоненциального соотношения выглядит так:
Ct = С0 • e_kelt, (ф. 4.2)
где Ct — концентрация в плазме в данное время (t), С0 — вычисленная начальная концентрация в плазме при t = 0, kel — константа скорости элиминации, е — основание натурального логарифма. Если взять логарифм концентрации в плазме, экспоненциальный процесс выглядит в виде прямой линии (рис. 4.11). Наклон линии на рис. 4.11 в действительности составляет kel/2,303. Деление на 2,303 необходимо потому, что концентрация в плазме дана в десятичном логарифме, а не в логарифме с основанием е. Такое представление использовано на рис. 4.11, т.к. десятичное основание более знакомо фармакологам. Если начальную экстраполированную концентрацию лекарства (С0) в компартменте разделить на введенную дозу, получают теоретический объем, необходимый для описания дозы лекарства. Это пространство называют условным объемом распределения (Vd).
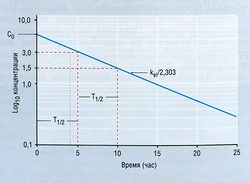
В то время как константа скорости элиминации показывает, как быстро элиминируется лекарство, расчет с использованием kel позволяет определить Т1/2, т.е. время, в течение которого концентрация лекарства в плазме падает на 50%. Поскольку падение концентрации лекарства со временем в логарифмическом исчислении является линейным, величина Т1/2 будет постоянной независимо от концентрации лекарства. Отношение kel и Т1/2 описывает уравнение:
Т1/2 х kel = 0,693 (ф. 4.3)
Поскольку величина T1/2 связана с Vd, значение Т1/2 не всегда отражает способность организма элиминировать лекарство. Предпочитаемый кинетический термин, показывающий способность организма удалять лекарство из кровотока, — это клиренс плазмы (Clp), равный Vd X kel. Клиренс остается постоянным для большинства лекарств, если механизмы элиминации не изменяются под влиянием патологических процессов и/или физиологических факторов.
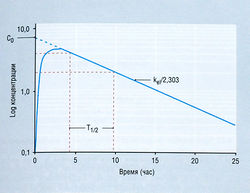
На рис. 4.11 показано снижение концентрации лекарства в плазме после его в/в введения. Однако при пероральном приеме необходимо время для абсорбции, и кривая снижения концентрации напоминает картину, изображенную на рис. 4.12. Форма кривой соотношения концентрации лекарства в плазме и времени отражает взаимодействие двух процессов первого порядка: поступления (абсорбции) вещества в одиночный кинетический компартмент и исчезновения (элиминации) вещества из него. Расчет Vd, Т1/2, kel и Сlр тот же самый, как после в/в введения, однако некоторые из производных параметров (Vd и Сlр) будут возрастать, если происходит значительная потеря лекарства до абсорбции (предсистемная элиминация вещества). Эта ситуация возникает в результате того, что концентрация вещества в плазме уменьшается вследствие предсистемной элиминация вещества, поэтому определение инициальной концентрации лекарства (С0) дает сниженную величину. Эта простая модель единственного однородного компартмента с кинетикой первого порядка вполне пригодна для расчета режима дозировки большинства лекарств.
Двухкомпартментная модель [ править | править код ]
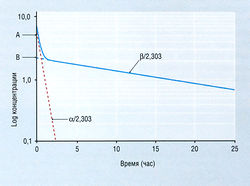
Для некоторых лекарств график, изображающий соотношение логарифма концентрации в плазме и времени, имеет криволинейную форму (рис. 4.13). Чтобы объяснить это явление, требуется расширить однокомпартментную модель. В простейшем виде организм следует рассматривать как двухкомпартментную модель (рис. 4.14). С определенным основанием можно считать, что объяснить криволинейный график на рис. 4.13 можно двумя линейными процессами. Эти два процесса известны под названием а- и β-фаз и характеризуются соответствующими для каждой из них константами скорости элиминации и Т1/2. Согласно этой модели, константа скорости кβ конечной линейной фазы (β) не та, что у kel на рис. 4.11 (модель одного компартмента). β-Фаза представляет собой более медленный процесс. Чем больше расхождение между кβ и kel, тем больше ошибка, если считать правомерной модель одного компартмента. К счастью, для большинства лекарств расхождение между кβ и kel не так велико, как межиндивидуальные различия кинетики, поэтому открытая модель одного компартмента служит приемлемой клинической аппроксимацией для индивидуального выбора дозировки.
Очевидно, что как одно-, так и двухкомпартментные модели дают чрезмерно упрощенное представление о распределении лекарств в организме, где лекарственные вещества абсорбируются и удаляются посредством метаболизма и экскреции. Когда расхождения в модельном распределении лекарств становятся клинически очевидными для данного лекарства, его применение следует рассматривать как особый случай более сложного соотношения распределение-эффект. Лекарства, подвергаемые дозозависимому распределению (например, фенитоин, аспирин), относятся к этой специальной категории более сложного соотношения между дозой, концентрацией и фармакологическими эффектами.

Описание к Рис. 4.13 Изменение концентрации лекарства в плазме в зависимости от времени после одномоментного внутривенного введения: открытая двухкомпартментная фармакокинетическая модель. Константа скорости конечной фазы III отличается от kel, и снижение концентрации со временем отражает более сложное соотношение между распределением и элиминацией лекарства. Начальное более быстрое падение концентрации циркулирующего лекарства (а) отражает в основном его перераспределение в периферический компартмент (Vp) (см. рис. 4.14) плюс незначительный компонент элиминации. Конечная, выраженная прямой линией фаза, отражающая зависимость концентрации в плазме от времени, представляет собой композит элиминации лекарства, замедленной возвратом вещества из Vp в центральный компартмент, в котором вещество распределяется быстро (Vc); тем самым снижается видимая скорость удаления лекарства из плазмы. Эту фазу обозначают как фазу кр. Обратная экстраполяция на время «ноль» дает отрезок, отсекаемый на координатной оси в точке В. Экстраполированное значение кр вычитают из наблюдаемой концентрации лекарства в то же самое время после его введения и откладывают на графике оставшуюся величину. Линия регрессии, проведенная через эти точки, дает наклон к„ и отрезок, отсекаемый на координатной оси в точке А, отражающий распределение лекарства в Vp.
Описание к Рис. 4.14 Схема открытой двухкомпартментной фармакокинетической модели, отражающая концентрации лекарства после внутривенного введения. При одномоментном (болюсном) в/в введении величина к„ несущественна, и в модели ею можно пренебречь. Если лекарство вводят путем инфузии, ка представляет собой константу нулевого порядка, равную скорости инфузии вещества. Существует центральный компартмент, в котором лекарство распределяется быстро (Vc) и из которого оно элиминируется, и периферический компартмент, в котором лекарство может распределяться (Vp), а затем возвращаться, выравнивая изменения концентрации вещества в центральном компартменте, когда лекарство элиминируется. kel — константа элиминации.
Описание к Рис. 4.15 Уравнения фармакокинетического распределения лекарства в отсутствие компартментализации. Константа конечной скорости распределения здесь обозначена А,, и ее интерпретация отличается от таковой для двух моделей, представленных на рис. 4.10-4.14. Введен новый расчет, называемый площадью под кривой (ППК), соответствующей соотношению между концентрацией в плазме и временем. С1р — клиренс плазмы; Т1/2 — период полувыведения; Vd — условный объем распределения.
Продолжительность действия лекарств
Подход без использования компартментного принципа [ править | править код ]
Этот подход используют для упрощенного определения фармакокинетических параметров, на основании которых можно рассчитать дозу лекарства. Многое в этом подходе заимствовано из открытой однокомпартментной модели. Площадь под кривой (ППК) концентрации представляет в интегральном виде соотношение между концентрацией в плазме и временем, и расчет кинетических параметров проводят соответственно уравнениям, приведенным на рис. 4.15. Этот метод до некоторой степени компенсирует экстраполяцию концентрации лекарства на С0 в модели одного компартмента, когда лекарство еще не абсорбировалось в организме после перорального приема.
Расчет дозировки [ править | править код ]
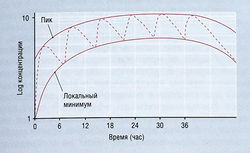
Большинство лекарств применяют длительно. Для лекарств с элиминацией первого порядка общее количество вещества в организме возрастает, пока экскретируемое количество не становится равным введенной дозе в расчете на единицу времени, т.е. концентрация в плазме достигает стационарного состояния. Время достижения этого состояния для таких лекарств зависит только от конечного Т1/2. Расчет показывает, что 94 и 97% стационарного состояния создаются через 4 и 5 Т1/2 соответственно. Для практических целей принимают, что стационарное состояние в это время существует (рис. 4.16). Чем чаще вводят дозу лекарства, тем выше его количество в организме при стационарном состоянии и тем меньше вариация между концентрацией в плазме на пике и минимальной концентрацией. Чем реже вводят лекарство, тем ниже его количество в организме при стационарном состоянии и тем больше различие между концентрацией в плазме на пике и минимальной концентрацией. Если интервал между введениями больше, чем 2 Т1/2, накопление лекарства при длительном приеме внутрь считается клинически несущественным.
Для быстрого создания терапевтической концентрации лекарства в плазме можно ввести ударную дозу (дозу насыщения)
Ударную дозу лекарства рассчитывают на основании его Vd и желаемой концентрации в плазме при стационарном состоянии (Css):
Ударная доза (мг/кг) = = Vd (л/кг) X Css (мг/л) (ф. 4.4)
При в/в введении ударную дозу обычно инфузируют в течение короткого периода времени, чтобы снизить риск возникновения побочных эффектов, связанных с очень высокой концентрацией лекарства в кровотоке. Затем рассчитывают величину поддерживающей дозы, основываясь на Сlр и величине интервалов между дозами (t):
Поддерживающая доза (мг/кг) = Сlр (л/кг/час) X Css (мг/л) X t (час) (ф. 4.5)
Нелинейная фармакокинетика
Нелинейная фармакокинетика [ править | править код ]
Нелинейная фармакокинетика (при которой клиренс, объем распределения и Т1/2 зависят от дозы или концентрации препарата) обычно обусловлена насыщением участков связывания на белках, насыщением ферментных систем печени, отвечающих за метаболизм лекарственного средства, или насыщением систем активного транспорта препарата в почках.
Насыщение участков связывания на белках [ править | править код ]
По мере роста молярной концентрации лекарственного средства участки связывания на белках насыщаются, и концентрация свободного препарата возрастает. Как правило, насыщение происходит, когда сывороточная концентрация находится в диапазоне от нескольких десятков до нескольких сотен микрограммов в миллилитре. Если лекарственное средство метаболизируется в печени и характеризуется низким отношением внутреннего печеночного клиренса к коэффициенту экстракции, при насыщении участков связывания на белках плазмы объем распределения и клиренс возрастают, а Т может оставаться неизменным (уравнение 1.12). В таких случаях зависимость между скоростью введения и средней концентрацией в стационарном состоянии нелинейна. Если же отношение внутреннего печеночного клиренса к коэффициенту экстракции велико, средняя концентрация в стационарном состоянии может линейно зависеть от скорости введения препарата. Печеночный клиренс при этом не зависит от связывания с белками плазмы, а увеличение объема распределения сопровождается ростом Т1/2 вследствие уменьшения скорости доставки препарата в печень. Большинство лекарственных средств характеризуются промежуточными значениями указанного отношения, поэтому предсказать изменение фармакокинетики при насыщении участков связывания на белках бывает трудно.
Насыщение систем элиминации [ править | править код ]
Для описания нелинейной (дозозависимой) элиминации обычно используют уравнение 1.2. Активные процессы всегда насыщаемы, но если концентрация препарата намного меньше Кm, фармакокинетику препарата можно считать линейной. Если же эта концентрация больше, чем Кm, необходимо использовать нелинейную модель. Насыщение систем элиминации и насыщение участков связывания на белках приводят к противоположным последствиям и, следовательно, могут компенсировать друг друга. В таком случае динамика концентрации препарата (например, салициловой кислоты в определенном диапазоне концентраций) описывается с помощью линейной модели.
При насыщении реакций метаболизма биодоступность препаратов, которые метаболизируются до поступления в системный кровоток, снижается в меньшей степени, и средняя концентрация препарата в стационарном состоянии возрастает непропорционально увеличению скорости введения. Продемонстрируем это, подставив уравнение 1.2 в уравнение 1.1 и решив полученное уравнение относительно Ссредн:
Когда скорость введения приближается к максимальной скорости элиминации (Vm), знаменатель в уравнении 1.15 стремится к нулю, а Ссредн непропорционально возрастает. На объем распределения насыщение реакций метаболизма не влияет; поэтому клиренс и скорость элиминации с ростом концентрации препарата уменьшаются, и зависимость концентрации от времени в полулогарифмических координатах представляет собой вогнутую кривую. Когда концентрация снизится до значения, при котором насыщения не происходит, эта зависимость примет вид прямой линии (как и в тех случаях, когда элиминация подчиняется кинетике первого порядка). T1/2 — величина постоянная, и потому этот показатель неприменим к лекарственным средствам, метаболизм которых насыщается в пределах терапевтического диапазона. Изменять дозу или частоту введения таких препаратов сложно, так как новое стационарное состояние достигается медленнее, а средняя концентрация в стационарном состоянии изменяется непредсказуемо.
Примером препарата, метаболизм которого насыщается в пределах терапевтического диапазона, служит фенитоин (Приложение II). Величина Кm для него (5—10 мг/л) близка к нижней границе терапевтического диапазона (10—20 мг/л). У некоторых больных (особенно у детей) Кm может составлять всего 1 мг/л. Требуемая сывороточная концентрация фенитоина — 15 мг/л — достигается при приеме препарата в дозе 300 мг/сут. Согласно уравнению 1.15, Vm = 320 мг/сут. При уменьшении дозы на 10% (270 мг/сут) Ссредн составит 5 мг/л, что значительно ниже требуемой. При увеличении же дозы на 10% (330 мг/сут) максимальная скорость элиминации (320 мг/сут) будет превышена на 10 мг/сут, и сывороточная концентрация фенитоина будет медленно, но неуклонно возрастать вплоть до развития побочных эффектов. Обеспечить столь точное (до 10%) дозирование невозможно, поэтому в тех случаях, когда требуемая сывороточная концентрация препарата более чем в 10 раз превышает Кт, чередование неэффективности и побочных эффектов почти неизбежно. При назначении лекарственных средств с узким терапевтическим диапазоном и дозозависимой элиминацией необходимо тщательно следить за их фармакологическими эффектами и сывороточной концентрацией (см. ниже).
ФАРМАКОКИНЕТИКА
ФАРМАКОКИНЕТИКА (греческий pharmakon лекарство + kinetikos относящийся к движению) — составная часть фармакологии, изучающая закономерности всасывания, распределения, метаболизма и выделения лекарственных средств. Исследование этих закономерностей основано на математическом моделировании указанных процессов.
Как самостоятельный раздел фармакологии фармакокинетика сформировалась в 30-х годах 20 века, но рассматривалась в качестве сугубо академической дисциплины вплоть до середины 60-х годов, когда на основе фармакокинетических принципов была разработана математическая модель оптимального режима применения сульфаниламидов (см. Сульфаниламидные препараты). С этого времени фармакокинетика стала интенсивно развиваться в нескольких направлениях (разработка новых фармакокинетических моделей, анализ связи между фармакокинетическими параметрами: изучение закономерностей распределения существующих и внедряемых в практику лекарственных средств и определение оптимальных режимов их назначения; исследование проблем биологической доступности и др.)- В современных условиях опреределение фармакокинетических характеристик новых лекарственных веществ является важной частью их доклинического и клинического испытания.
Основные фармакокинетические процессы. Каждое лекарственное вещество подвергается в организме всасыванию, распределению и выделению. Подавляющее большинство лекарственных веществ подвергается в организме также метаболическим превращениям.
Всасывание лекарственных веществ осуществляется за счет разных механизмов. Так, липофильные вещества всасываются главным образом путем пассивной диффузии через мембраны (см. Мембраны биологические). Гидрофильные вещества с невысоким молекулярным весом (массой) проникают путем фильтрации через поры биол. мембран. Многие вещества всасываются за счет активного транспорта их молекул с помощью транспортных систем клеточных мембран. Вещества белковой природы всасываются, очевидно, путем пиноцитоза (см.).
Распределение лекарственных веществ оценивается фармакокинетическими методами преимущественно по экстрацеллюлярной жидкости, включающей плазму крови, цереброспинальную жидкость, внутриглазную жидкость и жидкое содержимое желудочно-кишечного тракта. В норме объем экстрацеллюлярной жидкости у человека весом 70 кг принимается равным 15 л при общем количестве воды в организме около 40 л. Общий объем экстрацеллюлярной жидкости увеличивается при выпотах в брюшную и грудную полости, отеках и т. п., что может отражаться на распределении лекарств в организме.
Распределение лекарственных средств в организме обеспечивается системой кровообращения. Равномерному распределению лекарств препятствуют мембраны органов, клеток и клеточных органелл. При переносе лекарственного средства через мембраны возможно его частичное связывание с ингредиентами биологических жидкостей по обе стороны мембраны. Существуют разные типы связывания лекарственных средств, отличающиеся по степени специфичности. Наиболее универсально связывание лекарств на поверхности белковых молекул, главным образом альбуминов (см.) крови. Оно происходит за счет гидрофобного взаимодействия и характеризуется быстрой обратимостью. В картине общего распределения препаратов их связывание с белками крови имеет двоякое значение. С одной стороны, оно может сопровождаться понижением концентрации активного препарата и в соответствии с этим ослаблением эффекта; с другой стороны, связывание способствует депонированию препарата и тем самым продлевает его пребывание в организме. Так, медленное выведение и значительная продолжительность эффекта сульфаниламидов длительного действия и доксициклина во многом обусловлено высокой степенью связывания этих препаратов с белками крови.
Известно также специфическое связывание лекарств некоторыми тканями. Так, хорошо растворимые в липидах вещества, например, барбитураты (см.), депонируются в жировой ткани. При выходе из наркоза или при диализе по поводу отравления барбитуратами проявляется феномен так наз. вторичного сна, развивающийся вследствие мобилизации этих веществ из жировых депо. Другим примером специфического депонирования лекарств у человека является накопление тетрациклинов (см.) в растущей костной ткани и дентине зубов.
Наиболее важным участком связывания лекарственных веществ являются специфические рецепторы (см. Рецепторы, клеточные рецепторы). В области специфического рецептора концентрация лекарственного средства значительно превышает его концентрацию в окружающей биол. жидкости, но ввиду относительно малого размера рецептора это связывание обычно практически не отражается на общей картине распределения препарата в организме.
Лекарственные средства могут выделяться из организма в неизмененном виде или в виде метаболитов. Препараты, слабо растворимые в липидах, выделяются преимущественно почками в неизмененном виде. Препараты, относительно хорошо растворимые в липидах, подвергаются в почках обратному всасыванию эпителием канальцев и поступают вновь в систему кровообращения. Такие вещества выделяются почками лишь после того, как они путем метаболических превращений образуют хорошо растворимые в воде (полярные) соединения. Метаболические превращения лекарств в организме условно делят на два вида процессов — биотрансформацию и конъюгацию. Под биотрансформацией подразумевают реакции (окисление, восстановление, гидролиз), при которых одна функциональная группа молекулы лекарственного средства превращается в другую или в неполярное соединение вводится полярная группа. К реакциям конъюгации относятся биосинтетические процессы соединения лекарственных средств с эндогенными веществами, например, с глюкуроновой, серной и уксусной кислотами, а также с а-аминокислотами или метильным радикалом.
Биотрансформация осуществляется в эндоплазматическом ретикулуме (см.) гепатоцитов и катализируется системой оксидаз (см.). Конъюгация происходит также преимущественно в печени, но вне эндоплазматической ретикулума — в митохондриях (см.) или в растворимой фазе. В результате биотрансформации и конъюгации повышается гидрофилъность (см.) лекарственных средств, в результате чего понижается степень их реабсорбции эпителием извитых канальцев почек. Глюкурониды (см.), кроме того, могут секретироваться желчью и кишечным эпителием. Таким образом, биологическое значение биотрансформации и конъюгации заключается в подготовке липидорастворимых лекарственных веществ к выведению из организма. При этом обычно происходит ослабление или наступает полная утрата фармакологической активности лекарственных веществ. Однако в процессе биотрансформации метаболиты некоторых препаратов могут становиться активнее исходных лекарств. Так, фтазин (см.) и фталазол (см.) в процессе метаболизма в организме образуют более активные молекулы норсульфазола (см.) и сульфапиридазина (см.). В ряде случаев в результате метаболизма лекарств могут образовываться и токсические продукты. Токсические метаболиты образуются, например, при биотрансформации изониазида (см.), парацетамола (см.), фуросемида (см.) и др.
Выведение лекарств из организма происходит не только через почки, но также со слюной и через легкие.
Выведение препаратов с желчью или через стенку желудочно-кишечного тракта можно рассматривать в качестве экскреции или инкреции в зависимости от того, выводится ли препарат с фекалиями или всасывается в кишечнике вновь. В последнем случае существует возможность того, что при вторичном прохождении через печень с кровью воротной вены некоторая часть препарата будет вновь выделяться с желчью. В итоге образуется так называемый гепатопортальный круг обращения препарата. Такой особенностью фармакокинетики отличаются дигитоксин (см.), рифампицин (см. Рифамицины) и ряд других препаратов.
Моделирование фармакокинетических процессов. Принципы моделирования основных процессов фармакокинетика можно рассмотреть на следующем примере, заимствованном из физической химии.
Сосуд, разделенный полупроницаемой мембраной на две части, заполняют жидкостью в равных объемах. Если вещество X растворено в камере А и может проникать через мембрану в камеру В (но не обратно), то скорость переноса молекул вещества X из камеры А в камеру В зависит в каждый данный момент от его количества (концентрации) в камере А. Из этого следует, что скорость переноса вещества X непостоянна. Если бы скорость переноса вещества X из камеры А в камеру В не зависела от его концентрации, то она выражалась бы уравнением элементарной алгебры:
Однако учитывая, что скорость переноса в каждый интервал времени (t, — tt) разная, уравнение записывают следующим образом: = (1) tj — l i
Естественно, что скорость в любой момент интервала времени (t, — f,) определяется тем точнее, чем меньше интервал. Это дает в пределе бесконечно малые величины, в связи с чем можно вывести дифференциальное уравнение следующего вида: = (2)
Решение уравнения (2) приводит к так наз. экспоненциальному уравнению: МдСО = М°е
М, (3) А в котором, как и в уравнении (2), k — константа переноса, М° — количество препарата в камере в начальный момент времени, t — время, е — основание натуральных логарифмов.
Кривая, соответствующая экспоненциальной функции (3), соответствует изменению скорости переноса в зависимости от количества вещества X или его мгновенной концентрации.
Приведенная физико-химическая модель переноса вещества X из камеры А в камеру В является одновременно простейшей одночастевой моделью фармакокинетики, в к-рой камера А принимается за целый организм, а камера В — за внешнюю среду, куда выводится препарат X. Данная модель применима только при том условии, что препарат вводится в организм одномоментно, то есть внутривенно.
При пероральном введении препарата модель значительно усложняется. Коэффициент пропорциональности k в фармакокинетической модели приобретает смысл важного параметра — константы элиминации, которую обозначают в виде индекса el.
На основании одночастевой модели (а также более сложных моделей) о фармакокинетике препарата можно судить не только по данным концентрации его в крови, но и по данным кумулятивной экскреции препарата с мочой, со слюной и др. Для одночастевой модели скорость кумулятивной экскреции выражается уравнением, близким к уравнению (2), но с обратным знаком: dM dt
Это уравнение описывает накопление вещества в камере В. Метод кумулятивной экскреции менее точен, чем метод изучения концентрации препарата в крови, так как он не дает возможности установить концентрацию препарата в каждый момент времени, а позволяет выявить лишь среднюю концентрацию за определенный отрезок времени. Однако практические удобства этого метода очевидны. Особое значение имеет параллельное определение концентрации препарата в моче и крови, являющееся одним из приемов проверки адекватности одночастевой модели фармакокинетики.
Одночастевая модель отражает только конечную фазу пребывания препарата в организме, но не касается его распределения между тканями и органами. В связи с этим ее используют для анализа относительно несложных фармакокинетических процессов. Более совершенны так называемые многочастевые (многокамерные) модели разного типа.
Если правые части дифференциальных уравнений, описывающих перенос препарата из камеры в камеру, а также его выведение, представляют собой выражение прямой, то есть кривой первого порядка, как в уравнениях (2) и (6), то такие уравнения являются линейными, а модель, которую они описывают, относится к классу линейных. В классических многокамерных линейных моделях деление организма на камеры производят формально на основе анализа кривой выведения препарата, а не на основе анатомо-физиологического деления организма. Так, если при логарифмическом преобразовании эмпирических данных концентрации препарата в крови только часть кривой удовлетворительно приближенно выражается прямой линией, то это свидетельствует о том, что одночастевая линейная модель неадекватна реальному процессу выведения препарата. В таких случаях следует проверить степень приближения опытных данных с помощью двухчастевой или трехчастевой линейных моделей.
Причиной ошибочного принятия двухчастевой модели за одночастевую чаще всего является недостаточное число проб, взятых в самом начале фармакокинетического исследования.
Для выявления трехчастевой модели особенно важны ранние пробы. Трудность выполнения этого требования, возможно, объясняет причину, по которой трехчастевая линейная модель в фармакокинетике используется относительно редко.
Хотя выделение второй и далее камер основывается на формальном анализе кривой выведения препарата, существует некоторое предпочтительное отнесение определенных частей организма к первой, второй и третьей камерам. Обычно подразумевают, что к первой камере условно относится жидкая часть крови, интерстициальная жидкость и богато васкуляризированные органы (сердце, мозг, легкие, печень, почки и эндокринные железы); ко второй камере — все остальные ткани и органы. При использовании трехкамерной модели ко второй камере условно относят мышечную ткань, а к третьей — костную и жировую ткани.
В связи с развитием вычислительной техники особенно распространенной стала двухчастевая линейная модель. Большое число лекарственных средств, ранее изученных в рамках одночастевой модели, было изучено повторно с применением двухчастевой модели. В итоге были опубликованы уточненные значения фармакокинетических параметров и даны уточненные рекомендации относительно режима лечения.
Правая часть уравнений (2) и (6) иногда соответствует какой-либо кривой второго или даже высокого порядка. Такие модели называются нелинейными. Наиболее распространенным уравнением фармакокинетики второго порядка является выражение: dC VmaxC
Km 4- С ’, где Km и Vtnax — константы уравнения: Михаэлиса — Ментен (см. Кинетика биологических процессов).
Правая часть этого уравнения соответствует гиперболе. Она не может быть приведена к виду прямой ни в натуральных числах, ни путем логарифмического преобразования. Приближение к прямой имеет место только при условиях, когда значения концентрации препарата очень велики или очень малы по сравнению со значением константы Кт. Согласно кинетике второго порядка протекают все процессы выведения лекарственных средств путем метаболизма.
Особым классом фармакокинетических моделей являются так называемые перфузионные (физиологические) модели. Они основаны на предположении о пропорциональной связи между скоростью обратимого переноса препарата между кровью и тканями и интенсивностью кровоснабжения последних. Такой подход позволяет придать камерам модели реальное физиологическое содержание. Общий вид уравнений перфузионных моделей отличается от вида уравнений классических моделей тем, что вместо констант Ki> вводятся параметры скорости кровотока через органы, константы уравнения Михаэлиса — Ментен (в случае описания метаболизма препаратов), а также другие физиологические и биохимические константы.
Фармакокинетические параметры зависят от состояния организма. На них влияет характер питания, климат, курение и другие факторы. Некоторые значения параметров генетически детерминированы, напр, скорость ацетилирования изониазида. Выведение препаратов у новорожденных происходит значительно медленнее, чем у взрослых. Но в течение первых лет жизни скорость элиминации резко возрастает, превышая скорость элиминации у взрослых. В пожилом возрасте скорость элиминации замедляется. Среди различных факторов, влияющих на процессы фармакокинетики, можно назвать и биоритмы (см. Биологические ритмы). Однако-они до настоящего времени изучались преимущественно с точки зрения фармакодинамики (см.)
Среди патологических состояний, влияющих на фармакокинетику лекарственных средств, прежде всего следует отметить нарушение выделительной функции почек (см.). Для многих лекарственных средств найдены эмпирические коэффициенты зависимости между показателем выделительной функции почек, с одной стороны, и величиной дозы и интервалов ее введения — с другой.
Значение фармакокинетики для клинической медицины определяется ее практическими приложениями, важнейшими из которых являются: установление зависимости между степенью эффективности препарата и уровнем его в крови и основанная на этом оптимизация режима лечения; определение биол. доступности лекарственных средств из готовых форм; изучение взаимодействия лекарственных средств на путях их всасывания, распределения и элиминации (см. Несовместимость лекарственных средств). Знание Ф. каждого из применяемых в клинике лекарственных средств является одной из основ рациональной тактики лекарственного лечения (см. Фармакотерапия).



