Локализованная склеродермия: диагностика, клиника, лечение
В статье приводятся сведения о локализованной склеродермии, рассматриваются вопросы распространенности, этиопатогенеза, значения иммунных нарушений в реализации заболевания. Изложены клинические проявления локализованной склеродермии. Приведены данные диа
The article presents information on localized scleroderma, the issues of prevalence, etiology and pathogenesis, immune disorders values in the implementation of the disease, сlinical manifestations of localized scleroderma, diagnosis, prognosis and treatment of patients.
Подавление активности супероксиддисмутазы не «смягчает» реактивные соединения кислорода, способствующие фиброзированию. При ограниченной склеродермии наблюдаются также нарушения метаболизма коллагена и изменения в эндотелии сосудов. У взрослых женщин определенную роль в некоторых случаях играет микрохимеризм, то есть персистенция эмбриональных клеток после беременности. У 12% детей с ограниченной склеродермией положителен семейный анамнез на ревматические и аутоиммунные заболевания. Тот факт, что линейные формы склеродермии развиваются по линиям Блашко, свидетельствует о соматической мозаичности, подчеркивая тем самым эмбриональный генез или генетическую предрасположенность к заболеванию. Прежняя гипотеза о связи ограниченной склеродермии с хронической боррелиозной инфекцией не подтвердилась [4].
Больные могут предъявлять жалобы на зуд, болезненность, чувство покалывания и стянутости кожи, ограничение движений в суставах, изменение объема и деформацию тканей в пораженных участках тела [1]. Очаги склеродермии в своем развитии проходят три стадии: эритемы и отека, склероза (уплотнения) и атрофии кожи. В типичных случаях заболевание начинается с появления на коже розовых, розовато-сиреневых, ливидных или гиперпигментированных пятен округлой и/или полосовидной формы, иногда с явлениями отека. В стадию склероза из пятен образуются очаги уплотнения и утолщения кожи цвета слоновой кости с гладкой поверхностью и характерным восковидным блеском. По периферии очагов часто наблюдается воспалительный венчик лилового или розовато-фиолетового цвета, являющийся показателем активности процесса. В местах поражения кожа плохо собирается в складку, потоотделение уменьшено или отсутствует, нарушаются функция сальных желез и рост волос. С течением времени уплотнение кожи может уменьшаться. В стадию атрофии в очагах склеродермии развивается атрофия кожи, появляются телеангиэктазии, стойкая гипер- или гипопигментация. Кроме кожи в патологический процесс могут вовлекаться подлежащие ткани: подкожная клетчатка, фасции, мышцы, кости, реже — другие ткани и органы. Возможно спонтанное разрешение заболевания [1].
Ограниченная склеродермия подразделяется на лимитированную, генерализованную, линейную и глубокую формы.
Лимитированные формы ограниченной склеродермии
Бляшечная склеродермия характеризуется появлением на туловище или конечностях очагов эритемы и индурации кожи [1]. Типичны медленно растущие, одиночные или множественные округлые очаги диаметром от одного до нескольких сантиметров. Ранние очаги представляют собой нечетко ограниченные бледно-эритематозные или сиреневатые бляшки, распространяющиеся по периферии и не вызывающие субъективных жалоб. Центр очагов постепенно светлеет, приобретая оттенок цвета слоновой кости, и все больше затвердевает, так что в итоге очаг окружает только сиреневатый ободок («лиловое кольцо») (рис. 1). Процесс склерозирования продолжается, пока не сформируется блестящий атрофический пласт. Бляшки располагаются в основном на туловище, особенно в участках тесно прилегающей одежды, но встречаются и на конечностях, шее и лице. Они разрешаются в течение месяцев, оставляя коричневатую поствоспалительную гиперпигментацию [2].
.jpg)
Буллезная склеродермия характеризуется появлением в области очагов индурации и склероза кожи прозрачных пузырей, нередко сопровождающихся геморрагиями.
Узловатая (келоидоподобная) склеродермия характеризуется образованием на коже единичных или множественных узелков или узлов, внешне напоминающих келоидные рубцы. Очаги поражения развиваются, как правило, у больных, не имеющих склонности к развитию келоидов; их появление не связано с предшествующей травмой. Высыпания имеют телесный цвет или гиперпигментированы; излюбленная локализация — шея, туловище, верхние конечности. Буллезные или узловатые келоидные формы бляшечной склеродермии в детском возрасте встречаются очень редко.
Каплевидная склеродермия представляет собой мелкопятнистый тип заболевания, часто сочетается со склероатрофическим лишаем и встречается реже, чем бляшечный тип.
Идиопатическая атрофодермия Пазини–Пьерини многими авторами считается абортивным вариантом ограниченной склеродермии и клинически проявляется длительно существующими, незначительно западающими очагами коричневого или серо-коричневого цвета с фиолетово-сиреневым оттенком без признаков уплотнения кожи. Очаги располагаются чаще всего на туловище и верхних конечностях, часто симметрично или сегментообразно (рис. 2) [1].
.jpg)
Генерализованные формы склеродермии
При генерализованной склеродермии наблюдается появление множественных очагов эритемы и индурации кожи, занимающих несколько областей тела и нередко сливающихся в обширные очаги поражения [1].
Генерализованная склеродермия. Появление нескольких (> 3) отдельных очагов в различных анатомических участках.
Пансклеротическая склеродермия («инвалидизирующая пансклеротическая морфеа»): самая редкая и обширная форма склерозирования, является наиболее тяжелой формой заболевания, при которой поражаются все слои кожи и подлежащих тканей вплоть до костей, часто формируются контрактуры суставов с деформацией конечностей и длительно существующие язвы [1]. Это заболевание встречается преимущественно у детей препубертатного возраста. Вследствие быстро прогрессирующего обширного поражения дермы, подкожно-жировой ткани, мышц и фасций, а также костей в области туловища, конечностей (за исключением акральных участков) и лица развивается прогрессирующий склероз по типу панциря и, как следствие, контрактуры и рестрикция дыхательных движений. Рост тела ограничен. У детей часто развивается кахексия. Однако синдром Рейно или дисфагия, как при прогрессирующем системном склерозе, не наблюдаются [2].
Эозинофильный фасциит (синдром Шульмана). Проявляется в виде болезненного эритематозного и уплотнения проксимальных конечностей. Заболеванию обычно предшествует механическая травма. В отличие от других форм склеродермии чаще встречается у мальчиков. Склерозирование затрагивает глубокие фасции и сухожилия и может осложняться синдромом карпального канала. Только в 20% случаев наблюдаются типичные склеродермические изменения кожи. Характерным признаком является периферическая эозинофилия [2].
Линейные формы склеродермии являются самыми распространенными в детском возрасте формами проявления ограниченной склеродермии и подразделяется на варианты линейной и склеродермии по типу «удар саблей».
При линейной склеродермии на коже формируются полосовидные очаги эритемы и склероза, локализующиеся, как правило, на одной половине тела или по ходу нервно-сосудистого пучка. Очаги напоминают изменения при бляшечной склеродермии, однако сиреневая окраска проявляется не как кольцо, а находится на участке «активного» края, от которого распространяется очаг. Уплотненные, вытянутые в длину бляшки плотно спаяны с подлежащими тканями. Сморщивание более глубоких тканей (подкожно-жировой, фасций мышц, костей) приводит к прогрессирующей годами утрате массы вещества, следствием чего являются ограничение подвижности и контрактуры. Линейная склеродермия обычно бывает односторонней и развивается по линиям Блашко [6]. Описаны также двусторонние очаги и поражение одной половины тела. Чаще всего поражаются конечности (ноги чаще рук); реже сообщалось о проявлениях в области передней части грудной клетки, брюшной полости и ягодиц. Обычно очаги линейной склеродермии располагаются параллельно оси конечностей; реже встречаются поперечные или круговые очаги.
Склеродермия по типу «удар саблей». Очаг представляет собой полосовидный склеротический очаг цвета слоновой кости, в oбласти активного края которой в ходе заболевания развивается поствоспалительная гиперпигментация. Такие полосы выглядят впалыми, как будто плотно сросшимися с кожей головы. В области волосистой части кожи головы развивается рубцовая алопеция, иногда после осветления волос. Участок атрофии может распространяться на щеки, нос, верхнюю губу и иногда также в полость рта, поражая челюстные кости, в результате чего нарушается позиция зубов. Нередко уже в первый год заболевания развивается и ипсилатеральная асимметрия лица. В области очага могут быть затронуты и другие кости черепа. Нередко отмечаются неврологические симптомы, в частности, головные боли, нарушения способности к обучению или эпилептоидные припадки, анатомическим эквивалентом которых являет ипсилатеральный глиоз и признаки периваскулярного воспаления. Глаз на пораженной стороне также может быть вовлечен в патологический процесс, что проявляется энофтальмом, глазодвигательными нарушениями, изменениями радужки и глазного дна
Прогрессирующая гемиатрофия лица (синдром Парри–Ромберга) характеризуется прогрессирующей атрофией половины лица, проявляющейся преимущественно дистрофическими изменениями кожи, подкожной клетчатки, мышц и костей лицевого скелета. В отличие от распространенной склеродермии по типу «удар саблей», склерозирование кожи для синдрома Парри–Ромберга не характерно. Возможны переходы и одновременное наличие очагов линейной и других проявлений ограниченной склеродермии, а также с различными неврологическими нарушениями, включая эпилепсию [1].
Глубокие формы склеродермии. В редких случаях процесс склерозирования затрагивает не столько дерму, сколько подкожно-жировую ткань или еще глубже расположенные участки. Такие очаги, в отличие от бляшечной и линейной формы, не имеют четких границ. Различают следующие формы проявления глубокой склеродермии:
В случае линейных и глубоких форм склеродермии, при поражении мышц или при локализации вблизи суставов могут образоваться контрактуры. Наблюдаются также ипсилатеральные неврологические изменения (головная боль, изменения на электроэнцефалограмме, приступы судорог). В случае гемиатрофии лица в дополнение к аномалиям расположения зубов могут развиться параличи глазных мышц [2].
Локализованную склеродермию следует дифференцировать со следующими заболеваниями: системная склеродермия и другие диффузные болезни соединительной ткани, склередема Бушке, склеромикседема, диффузный эозинофильный фасциит Шульмана, склеродермоподобная форма базально-клеточного рака кожи, хронический атрофический акродерматит, индуцированные склеродермоподобные заболевания, вызванные применением лекарств и пищевых добавок (блеомицин, витамин К, L-триптофан), использованием силиконовых протезов, контактом с химикатами (хлорвинил, органические растворители) и пр., келоидные и гипертрофические рубцы, склеродермоподобная форма хронической болезни «трансплантат против хозяина», липодерматосклероз, липоидный некробиоз, поздняя кожная порфирия, саркоидоз, амилоидоз, синдром Вернера, фенилкетонурия, радиационный фиброз, панникулит, псевдопелада Брока, соединительнотканный невус, кольцевидная гранулема и др. [1, 7].
Несмотря на то, что в отличие от системной склеродермии специфические лабораторные параметры для ограниченных форм склеродермии отсутствуют, многие параметры могут быть полезны при дифференциальной диагностике. Это, в частности, относится к ранней фазе заболевания и к вариантам линейной склеродермии детского возраста. Следует также помнить, что ни какие из серологических параметров не подходят для оценки активности заболевания, контроля в динамике или оценки эффективности терапии. В ходе заболевания ведется клинический мониторинг, по показаниям с помощью сонографии (20 МГц). Детям с синдромом Парри–Ромберга и всеми формами глубокой склеродермии показано обследование с использованием методов визуализации, в частности магнитно-резонансной томографии (МРТ), для оценки масштаба поражения подкожных структур. При глубоких формах склеродермии возможны и внутримозговые изменения. Для дермы и подкожно-жировой клетчатки подходит высокочастотное (20 МГц) ультразвуковое исследование. Результаты сонографии и МРТ, а также параметры толщины кожи, измеренные дюрометром, служат маркерами активности заболевания [2, 8].
Поскольку клиническая картина заболевания типична, гистологическое исследование обычно не требуется. Для гистологической картины на ранних стадиях заболевания характерны дермальный отек, периваскулярная лимфоцитарная инфильтрация и дегенерация коллагеновых волокон. В дальнейшем течении заболевания происходит утолщение дермы, уменьшение эластических волокон и уплотнение волокон коллагена. Придатки кожи и подкожно-жировая клетчатка вытесняются содержащей гиалин соединительной тканью. В случае глубоких форм морфеа биопсия также должна проводиться на соответствующую глубину, то есть вплоть до фасций [9, 10].
Лечение каждому больному необходимо подбирать индивидуально в зависимости от формы, стадии и тяжести течения заболевания, а также локализации очагов поражения, особенно ввиду высокой степени спонтанной ремиссии бляшечных форм заболевания. Целью терапии является предотвращение дальнейшего развития склерозирующего воспаления. Показания к терапии имеются, как правило, при наличии признаков активного воспаления, на образовавшийся рубец терапевтически повлиять уже практически невозможно. Целесообразность назначения больным склеродермией высоких доз пеницилламина (750–1000 мг в сутки и выше) в настоящее время является спорной [11]. Основными для терапии ограниченной склеродермии являются следующие принципы.
Противовоспалительная терапия: хороший опыт имеется с местными глюкокортикоидами (классов II–III), местным кальципотриолом. Производные витамина D оказывают иммуномодулирующее действие (подавляют активацию и рекрутирование Т-клеток) и блокируют пролиферацию фибробластов.
Иммуносупрессивная терапия метотрексатом (0,3–0,6 мг/кг массы тела в неделю) и глюкокортикоидами (метилпреднизолон 1–2 мг/кг массы тела в день или пульс-терапия в дозе 30 мг/кг массы тела в течение трех дней в месяц).
При линейных формах склеродермии следует рано начинать системную терапию, чтобы избежать дальнейших последствий заболевания [1]. Кроме того, при распространенном поражении показаны физиотерапевтические мероприятия (лечебная гимнастика, мануальный лимфодренаж). Хирургические мероприятия целесообразны только в случае подтвержденной неактивной стадии заболевания. Это касается как ортопедических мероприятий (удлинение ахиллова сухожилия, эпифизеодез для выравнивания разницы в длине ног), так и мероприятий пластической хирургии (например, при гемиатрофии лица) [12, 13].
Перспективным подходом к лечению склеродермии с достижением дефиброзирующего эффекта является применение иммуномодуляторов (диуцифон, Ксимедон, Галавит, Ридостин и др.) [5, 14, 15].
Больным локализованной склеродермией специальной диеты не требуется. При поражении конечностей необходимо избегать чрезмерной физической нагрузки, резких движений, ударов и толчков.
Физиотерапевтическое лечение проводится под контролем врача-физиотерапевта.
Также применяют фонофорез, электрофорез гиалуронидазы и терапию лазерным излучением красного диапазона (длина волны 0,63–0,65 и 0,89 мкм). Лечебная гимнастика и массаж особенно рекомендуется больным линейной формой склеродермии при ограничении движений в суставах и формировании контрактур.
Хирургическая коррекция необходима при наличии сгибательных контрактур и деформаций тела (области головы, суставов, конечностей). При наличии косметических дефектов, в основном, у больных саблевидной формой локализованной склеродермии («удар саблей») и прогрессирующей гемиатрофией Парри–Ромберга показана пластическая хирургическая коррекция. Хирургические вмешательства необходимо проводить в неактивную стадию локализованной склеродермии (при отсутствии активности заболевания в течение нескольких лет). Больным склероатрофическим лихеном, локализующимся в области крайней плоти полового члена и вызывающим стойкий фимоз, в случаях отсутствия эффекта от консервативной терапии показана циркумцизия [1].
Критериями эффективности лечения являются прекращение прогрессирования заболевания, разрешение или уменьшение эритемы, отека, утолщения и уплотнения кожи, а также других симптомов склеродермии; устранение или уменьшение субъективных ощущений. Госпитализация необходима при развитии тяжелых форм локализованной склеродермии или осложнений заболевания, а также при отсутствии эффекта от амбулаторного лечения или условий для его проведения.
Больным локализованной склеродермией необходимо избегать чрезмерного солнечного облучения, травм, переохлаждения и перегревания, простудных заболеваний, стрессовых ситуаций, необоснованного применения лекарственных средств. Рекомендуется проводить санацию очагов фокальной инфекции. Необходимо динамическое наблюдение врача-дерматовенеролога с целью контроля за течением заболевания и раннего выявления симптомов диффузных болезней соединительной ткани. При сопутствующих заболеваниях необходимо наблюдение и лечение у соответствующих специалистов — терапевта, эндокринолога, гинеколога, гастроэнтеролога, невропатолога, оториноларинголога. При тяжелых формах необходима медицинская и социальная реабилитация больных (направление на ВТЭК и трудоустройство) [1].
Прогноз заболевания зависит от соответствующей формы склеродермии. Так, у 50% больных бляшечными формами процесс клинически регрессирует в течение примерно трех лет или, как минимум, наблюдаются признаки размягчения очагов. В случае глубоких форм это происходит в среднем через пять лет. После линейных и глубоких форм нередко остаются уродливые рубцы. Переход в прогрессирующий системный склероз происходит только в случае пансклеротической склеродермии [2].
Таким образом, локализованная склеродермия остается одной из наиболее актуальных проблем современной медицины в связи с широким ее распространением, вариабельным клиническим течением, заболеванием лиц наиболее трудоспособного возраста и рефрактерностью ко многим методам терапевтического воздействия. Индивидуально подобранный комплексный подход к лечению каждого больного в зависимости от формы, стадии и тяжести течения заболевания, а также локализации очагов поражения позволит предотвратить дальнейшее развитие склерозирующего воспаления.
Литература
* ГБОУ ДПО КГМА МЗ РФ, Казань
** ГАУЗ «Республиканский клинический кожно-венерологический диспансер» МЗ Республики Татарстан, Казань
Диагностика и лечение системной склеродермии
Системная склеродермия (ССД) – аутоиммунное заболевание, характеризующееся вовлечением в воспалительный процесс соединительной ткани и проявляющееся поражением кожи, внутренних органов, сосудов и суставов. В статье рассматриваются современные подходы к лечению ССД, которые изложены в недавно опубликованных рекомендациях Европейской антиревматической лиги (EULAR).
Системная склеродермия (ССД) – это аутоиммунное заболевание, в основе которого лежат генерализованная микроангиопатия и активация процессов фиброза кожи и внутренних органов. На ранних стадиях заболевание проявляется кожными изменениями в виде плотного отека пальцев и синдромом Рейно, которые могут не сопровождаться ухудшением общего состояния или признаками поражения внутренних органов (дисфагией, одышкой и др.), поэтому пациенты зачастую не сразу обращаются за медицинской помощью. В связи с этим ССД нередко диагностируют поздно, когда патологические изменения в органах необратимы, а лечение менее эффективно. По данным канадского регистра, у 408 пациентов диагноз ССД был установлен в среднем через 6,0 лет после развития феномена Рейно и через 2,7 года после появления первых “внекожных” проявлений [1]. В России ССД диагностировали через 2,0–2,7 года после появления феномена Рейно при диффузной форме заболевания и через 4,8–6,5 года при лимитированной форме, что связано с разной частой поражения внутренних органов, а также скоростью прогрессирования заболевания [2]. При этом результаты крупного исследования (n=5860) показали, что смертность пациентов с ССД достигает 68 на 1000 человек в год [3]. Таким образом, своевременная диагностика ССД представляет собой сложную, но очень важную задачу для врача.
В настоящее время ССД активно изучается в рамках проекта EUSTAR (European League Against Rheumatism Scleroderma Trials and Research Group). В Российской Федерации в нем принимают участие два центра – НИИ ревматологии им. В.А. Насоновой и клиника им. Е.М. Тареева.
Эпидемиология и факторы риска
Системной склеродермией женщины страдают чаще мужчин (3:1); большая часть пациентов находится в возрасте от 25 до 50 лет [4]. Заболеваемость отличается в разных регионах. Так, в Северной Европе и Японии она составляет менее 10 на 1 млн населения в год, а в Южной Европе, Северной Америке и Австралии достигает 14-21 на 1 млн в год [5]. Распространенность заболевания среди афроамериканцев, американских индейцев, австралийцев, японцев выше, чем среди европейцев и белого населения США [6].
Выявлено множество генов, участвующих в регуляции иммунной системы и повышающих риск развития ССД, в том числе BANK1, C8orf13-BLK, IL-23R, IRF5, STAT4, TBX21 и TNFSF4 [7]. Изучаются также потенциальные эпигенетические механизмы и роль факторов окружающей среды, в том числе кремниевой пыли, органических растворителей, лекарственных препаратов (блеомицина, карбидопы и др.), пестицидов, рапсового масла, кокаина [8].
Клиническая картина
Выделяют две основные формы ССД – диффузную и лимитированную. При лимитированной форме уплотнение кожи отмечается дистальнее локтевых и коленных суставов, при диффузной – изменения кожи могут быть выявлены на туловище, бедрах и плечах (поражение кожи лица встречается при обеих формах). Различия между двумя формами заболевания не ограничиваются распространенностью кожного процесса: диффузная форма характеризуется частым поражением внутренних органов и более быстрым прогрессированием заболевания. Если при диффузной форме ССД 10-летняя выживаемость составляет 65%, то при лимитированной она достигает 92% [9].
Феномен Рейно. Феномен Рейно встречается у 95% больных системной склеродермией и, как правило, оказывается первым признаком болезни [10]. Клинически он имеет две или иногда три стадии – побеление, цианоз и покраснение кожи пальцев, которые развиваются под действием холода и могут сопровождаться онемением и болью 11. Для первичного синдрома Рейно, в отличие от феномена Рейно при ССД, характерны отсутствие изменений при видеокапилляроскопии ногтевого ложа, антинуклеарных антител, симптомов ишемического повреждения тканей (гангрена, язвочки, рубчики), нормальное значение СОЭ [16].
Поражение кожи. Другим признаком ССД является поражение кожи, которое развивается в три стадии: отек (например, плотный отек кистей), уплотнение ( например, склеродактилия), атрофия. На первом этапе отмечаются плотный отек и снижение эластичности кожи и подлежащих тканей, в дальнейшем формируется “склеродерма”, а на стадии атрофии кожа истончается и приобретает цианотично-бурый цвет, появляется специфический блеск, исчезает волосяной покров [17]. Характерны симптом “кисета” (радиальные складки вокруг рта) (рис. 1) и увеличение количества телеангиоэктазий [18]. В результате поражения сосудов микроциркуляторного русла нередко наблюдается ишемическое повреждение кожи – рубчики на дистальных фалангах пальцев по типу “крысиных укусов”, реже – сухие некрозы или гангрена [19]. Существуют и другие специфичные для ССД типы поражения кожи, такие как гипо- и гиперпигментация (“salt and pepper”), кожный кальциноз [20].
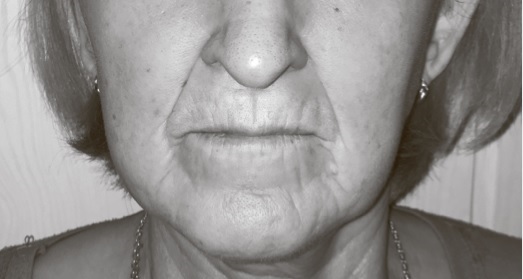 Рис. 1. Кисетный рот у пациентки с ССД
Рис. 1. Кисетный рот у пациентки с ССД
Для оценки выраженности кожных изменений применяют модифицированный кожный счет по Rodnan, предполагающий определение толщины кожного покрова в 17 участках тела в баллах от 0 до 3 (нормальная толщина, незначительное, умеренное и выраженное утолщение) с расчетом суммарного балла (максимальное значение 51) [21]. Другим методом, используемым для анализа степени повреждения кожи, является ультразвуковое исследование: гиперэхогенность расценивается как уплотнение кожи вследствие избыточного отложения коллагена, а гипоэхогенность – как признак отека тканей [22]. Реже в клинической практике используют дюрометрию – измерение твердости кожи [23].
Поражение внутренних органов. В России очень большой вклад в изучение висцеральных проявлений ССД внесла проф. Н.Г. Гусева, которая работала в клинике, возглавляемой Е.М. Тареевым, а затем в НИИ ревматологии под руководством акад. В.А. Насоновой [24,25].
У большинства пациентов с ССД (70-98%) развивается поражение желудочно-кишечного тракта, в частности гипотония пищевода, которая проявляется дисфагией и гастро-эзофагеальным рефлюксом. Возможно развитие синдрома мальабсорбции и избыточного роста патогенной флоры на фоне замедления пассажа химуса, а также поражение толстой кишки (диарея, недержание кала) [26]. В отечественном исследовании установлена взаимосвязь между гастроэзофагеальным рефлюксом и тяжестью легочного фиброза [27].
Поражение сердца встречается в 15-35% случаев [28,29] и проявляется сердечной недостаточностью, нарушениями ритма, болевым синдромом [30]. В редких случаях развиваются пороки клапанов, в том числе митральный стеноз [31].
Примерно у 75% больных уже в первые годы заболевания выявляют интерстициальное поражение легких, характеризующееся медленным прогрессирующим течением с исходом в фиброз разной степени тяжести [32]. Л.В. Теплова и соавт. с помощью компьютерной томографии высокого разрешения выявили признаки поражения интерстиция легких у 82% из 138 пациентов с ССД [33].
Для ССД характерно развитие легочной артериальной гипертензии (ЛАГ), иногда тяжелой. По данным недавно опубликованного исследования, в течение в среднем 4 лет умерли 60 из 132 пациентов с ССД, осложнившейся ЛАГ. Выживаемость от момента установления диагноза ЛАГ составила всего 4 (2,2-6,2) года [34]. Легочная гипертензия у пациентов с ССД может быть следствием следующих состояний: ЛАГ (в том числе как исход отложения коллагена в стенке сосудов), легочной веноокклюзионной болезни и/или легочного капиллярного гемангиоматоза, дисфункции левого желудочка, поражения легких с гипоксемией, хронической тромбоэмболической ЛГ [35,36].
Поражение почек встречается у 19% пациентов [29]. Частота развития острого склеродермического криза, проявляющегося быстрым ухудшением функции почек (острое почечное повреждение), составляет 10-15% при диффузной форме ССД и 1-2% при лимитированной форме [37,38]. В настоящее время поражение почек не является основной причиной гибели больных, уступив легочному фиброзу. Склеродермический почечный криз может быть заподозрен при впервые выявленном повышении АД >150/85 мм рт. ст., сохраняющемся в течение последующих 24 ч, а также снижении скорости клубочковой фильтрации на 10% или измеренной СКФ менее 90 мл/мин. Дополнительными признаками склеродермического почечного криза могут быть впервые возникшая гематурия и протеинурия, внезапный отек легких, олигурия или анурия, ретинопатия [39].
Диагностика
ССД следует подозревать у всех пациентов с феноменом Рейно. Важное диагностическое значение имеет поражение кожи (уплотнение кожи, “кисетный” рот, маскообразное лицо, склеродактилия, кальциноз, пигментация), хотя оно может и отсутствовать. При опросе следует также обращать внимание на симптомы поражения внутренних органов, такие как одышку и дисфагию. Для диагностики ССД на практике используют классификационные критерии, разработанные экспертами Американской коллегии ревматологии (ACR) и Европейской антиревматической лиги (EULAR) в 2013 г. (табл. 1) [40]. Необходимо учитывать, что критерии ACR-EULAR мало информативны на ранней и очень ранней стадии ССД, в то время как результаты исследования EUSTAR показали, что срок между развитием синдрома Рейно и других симптомов ССД составляет в среднем 4,8 года при лимитированной форме и 1,9 года при диффузной форме [41]. Полагают, что лечение, начатое в этот период (так называемое “window of opportunity”, или “окно возможностей”), может предотвратить развитие поражения внутренних органов и замедлить прогрессирование заболевания. В связи с этим были разработаны критерии ранней диагностики системной склеродермии (VEDOSS; табл. 2) [42]. На первом этапе диагностики предлагается выявлять ключевые признаки заболевания (так называемые “красные флаги”), такие как синдром Рейно и плотный отек пальцев кистей. На втором этапе проводят видеокапилляроскопию ногтевого ложа и определяют специфические антитела (например, антицентромерные или к топоизомеразе-1) [43]. При очень ранней ССД поражение внутренних органов отсутствует, в то время как при ранней ССД отмечают признаки субклинического их поражения, например, диастолическую дисфункцию левого желудочка по данным эхокардиографии (уменьшение отношения E/A), начальное снижение диффузионной способности легких ТАБЛИЦА 1. Классификационные критерии системной склеродермииACR-EULAR 2013 г
Не стоит забывать о существовании ССД без склеродермы, при которой поражение кожи (уплотнение и фиброз) отсутствует как на ранних, так и поздних стадиях заболевания. В этом случае диагноз устанавливают на основании наличия синдрома Рейно, дигитальных язв, специфических антител, изменений при видеокапилляроскопии, висцеральных поражений [45,46]. Также выделяют CREST-синдром – сочетание кальциноза кожи, синдрома Рейно, нарушения моторики пищевода, склеродактилии и телеангиоэктазий, а также антител к центромере. Для СREST-синдрома характерно достаточно благоприятное течение [47].
| I. Выявление “красных флагов” (феномен Рейно, плотный отек пальцев кистей/склеродактилия) |
| II.а) Проведение видеокапилляроскопии ногтевого ложа (ранние, активные, поздние изменения) |
| б) Серологические исследования: выявление специфических аутоантител (anti-Scl70, ACA, anti-RNA pol III и др.) |
| III. Установление диагноза очень ранней системной склеродермии при выявлении изменений, соответствующих пунктам I и II. |
| IV. Дальнейшее обследование пациентов, соответствующих критериям очень ранней системной склеродермии (компьютерная томография органов грудной клетки, эхокардиография, исследование функции внешнего дыхания, манометрия пищевода). |
| V. Установление диагноза ранней системной склеродермии при выявлении изменений, соответствующих пунктам I, II, IV. |
Важным лабораторным критерием диагностики ССД является наличие аутоантител, например, антител к топоизомеразе I (anti-Scl-70), антицентромерных антител (ACA), антител к рибонуклеопротеазе III (anti-RNA pol III), anti-Th/To, anti-U3RNP/Fibrillarin, anti-Pm/Scl, anti-Ku, антител к белкам сплайсосомы (anti-U1RNP, anti-Sm). В отечественном исследовании (n=300) было показано, что у большинства пациентов с ССД определялись антинуклеарный фактор (83,8%) и anti-Scl-70 (50,0%), реже встречались ACA (14,6%), anti-U1RNP (8,6%) и anti-RNA pol III (5,5%) [48]. Различные типы аутоантител могут ассоциироваться с определенными клиническими проявлениями ССД (табл. 3), поэтому их исследование имеет не только диагностическое, но и прогностическое значение.
Важное диагностическое значение имеют изменения при видеокапилляроскопии. Условно выделяют следующие стадии изменений, выявляемых при видеокапилляроскопии ногтевого ложа у больных с ССД: раннюю, активную и позднюю. При склеродермическом типе изменений на разных стадиях могут быть обнаружены гигантские капилляры, капиллярные кровоизлияния, уменьшение количества капилляров или аваскулярные участки, дезорганизация архитектуры капиллярного русла, “ветвистые” капилляры [49].
Лечение
Международные рекомендации по лечению системной склеродермии были разработаны рабочей группой EULAR/EUSTAR в 2016 году [85]. Они подразделены в зависимости от проявлений заболевания (феномен Рейно, язвы дистальных фаланг пальцев, легочная гипертензия, поражение кожи и легких, склеродермический почечный криз, поражение желудочно-кишечного тракта).
Феномен Рейно. Пациентам с феноменом Рейно следует избегать нахождения на холоде и бросить курить. Препаратами первой линии считают дигидропиридиновые антагонисты кальция, например, пролонгиро ванные формы нифедипина или амлодипин. Благоприятное влияние препаратов этой группы на частоту и тяжесть эпизодов ишемии подтверждается результатами мета-анализа клинических исследований [50]. При недостаточной эффективности антагонистов кальция возможно применение ингибиторов фосфоди эстеразы 5 типа (например, силденафила), эффективность которых также была показана при мета-анализе клинических исследований. Однако они несколько чаще вызывают нежелательные явления, в том числе вазомоторные реакции, миалгии, боль в груди, диспепсию и нарушения зрения [51]. При тяжелом феномене Рейно возможно внутривенное введение синтетического аналога простациклина илопроста (0,5-3 нг/кг/мин в течение 3-5 дней) или ингаляционное его применение в дозе 50-150 мкг два раза в сутки. В Российской Федерации для лечения синдрома Рейно чаще при меняют синтетический аналог простагландина Е1 альпростадил. В неконтролируемых клинических исследованиях альпростадил вызывал уменьшение или купирование боли, ускорял заживление язв и улучшал показатели микроциркуляции [24,52]. В 1985 году было проведено многоцентровое, двойное слепое, плацебоконтролируемое исследование, в которое было включено 55 пациентов с первичным феноменом Рейно и феноменом Рейно в рамках ССД. 72-часовое внутривенное введение альпростадила улучшало кровоток в микроциркуляторном русле по сравнению с плацебо [53].
При непереносимости вазодилататоров в качестве альтернативного препарата в рекомендациях указан флуоксетин 20 мг/сут (селективный ингибитор обратного захвата серотонина), хотя он изучался только в одном небольшом исследованием (n=27) пациентов), в котором по эффективности превосходил нифедипин у пациентов с тяжелым феноменом Рейно [54].
Обсуждается возможность применения топических аналогов нитроглицерина для лечения феномена Рейно. Так, в многоцентровом, двойном слепом, плацебоконтролируемом исследовании MQX-503 (аналог нитроглицерина, быстро абсорбирующийся с поверхности кожи) улучшал микроциркуляцию у пациентов с феноменом Рейно [55]. Длительное время считалось, что блокаторы эндотелиновых рецепторов (бозентан) эффективны только в профилактике новых дигитальных язв, но мало влияют на течение феномена Рейно. Однако недавно было показано, что бозентан может применяться для лечения феномена Рейно у пациентов с ССД, которым противопоказана терапия простаноидами [56,57].
Дигитальные язвы. Пациентам с дигитальными язвами следует избегать нахождения на холоде и бросить курить. Основными препаратами считают простаноиды (например, внутривенное введения илопроста) и ингибиторы фосфодиэстеразы 5 типа, эффективность которых подтверждена результатами мета-анализов клинических исследований [58]. Из ингибиторов фосфодиэстеразы 5 типа у пациентов с дигитальными язвами лучше всего изучен тадалафил, в то время как результаты применения силденафила оказались достаточно противоречивыми. По данным одного из последних исследований SEDUCE, при его применении частота возникновения язвенных дефектов была даже выше, чем в группе плацебо [59]. Тем не менее, авторы рекомендаций отмечают, что доза силденафила в указанном исследовании была ниже (60 мг/сут), чем в других исследованиях (100 мг/сут). При неэффективности ингибиторов фосфодиэстеразы может быть назначен бозентан (неселективный антагонист ЕТА и ЕТВ эндотелиновых рецепторов), эффективность которого была установлена в двух рандомизированных исследованиях (RAPIDS-1, RAPIDS-2). В этих исследованиях лечение бозентаном вызывало уменьшение количества новых язвенных дефектов, но не способствовало заживлению уже имевшихся язв [56,60]. Бозентан может оказывать гепатотоксическое действие, а также обладает тератогенностью. При этом он снижает эффективность пероральных контрацептивов, ингибируя систему цитохрома Р450. Таким образом, ввиду скорее профилактического действия препарата и серьезности нежелательных явлений, бозентан считается препаратом третьей линии [61]. В острую фазу или при подозрении на тромботические осложнения к терапии могут быть присоединены антиагреганты (аспирин, клопидогрел) и антикоагулянты (эноксапарин, варфарин) [58,62,63]. При наличии некротизированной ткани в области ишемического дефекта необходима первичная хирургическая обработка раны, назначение антибактериальной терапии при инфицировании язвы, применение специальных повязок для улучшения заживления язвы 65. Для купирования болевого синдрома применяют нестероидные противовоспалительные препараты, опиоиды, лидокаин [62].
Легочная артериальная гипертензия. Применение антагонистов эндотелиновых рецепторов, таких как бозентан, амбризентан, мацитентан, приводит к повышению толерантности к физическим нагрузкам и замедляет прогрессирование легочной гипертензии [6769]. Зависимость эффекта от дозы была отмечена только при лечении амбризентаном (доза 10 мг по эффективности превосходила дозу 5 мг). К такому же эффекту приводило применение ингибиторов фосфодиэстеразы 5 типа, в том числе силденафила (от 60 до 240 мг/сут) и тадалафила (40 мг/сут), а также риоцигуата – стимулятора растворимой гуанилатциклазы [67,69,70]. При этом эффективность риоцигуата (улучшение пробы с 6-минутной ходьбой) была доказана у пациентов как с ЛАГ, так и с хронической тромбоэмболической легочной гипертензией в исследованиях PATENT и CHEST [71]. Более высокая эффективность комбинированной терапии бозентаном и силденафилом у пациентов с ЛАГ (в том числе идиопатической, наследственной и в рамках диффузных заболеваний соединительной ткани) не была подтверждена в исследовании COMPASS [72]. При тяжелой ЛАГ возможно внутривенное применение эпопростенола (стартовая доза 2 нг/кг/мин), которое в двух исследованиях приводило к улучшению толерантности к физическим нагрузкам и показателей гемодинамики [73,74]. Недостатком препарата является короткий период полувыведения, в связи чем его приходится вводить с помощью центрального венозного катетера, а также синдром отмены, характеризующийся резким повышением давления в легочной артерии при прекращении лечения. Альтернативой может служить ингаляционное применение илопроста, хотя следует учитывать, что его эффективность в лечении ЛАГ у пациентов с ССД недостаточно изучена [67,69]. Ранее пациентам с ЛАГ назначали терапию варфарином, однако в регистре COMPERA не была подтверждена эффективность антикоагулянтов у пациентов с ССД [75,76].
Поражение кожи. Ранние кожные изменения при ССД могут поддаваться лечению метотрексатом. В двух рандомизированных клинических исследованиях препарат изучался в дозах 15 мг/нед в течение 2 лет и 10 мг/нед в течение года. Эти дозы ниже доз препарата, которые применяются при других ревматических заболеваниях, в частности ревматоидном артрите [40,77]. Применение циклофосфамида у пациентов с висцеральными проявлениями ССД в рандомизированных клинических исследованиях также приводило к улучшению состояния кожи [78]. Эффективность микофенолата мофетила и азатиоприна изучена недостаточно.
Интерстициальная болезнь легких. В рандомизированном клиническом исследовании (Scleroderma Lung Study) лечение циклофосфамидом в дозе 1-2 мг/кг/сут внутрь вызывало уменьшение одышки и изменений на компьютерных томограммах, улучшение качества жизни и показателей вентиляционной функции, причем наиболее выраженный эффект был отмечен у пациентов с тяжелым поражением легких [78]. В другом многоцентровом, проспективном, рандомизированном, двойном слепом, плацебо-контролируемом исследовании циклофосфамид применяли внутривенно в дозе 600 мг/м2/мес в течение 6 месяцев, а затем заменяли на азатиоприн (2,5 мг/кг/сут) в течение 6 месяцев. В этом исследовании был выявлен прирост форсированной жизненной емкости (ФЖЕЛ) легких в основной группе на 4,2% по сравнению с контролем [79]. Был сделан вывод, что циклофосфамид замедляет прогресси ро вание заболевания, а его применение оправдано у пациентов с прогрессирующим интерстициальным поражением легких. Режим дозирования и длительность терапии следует подбирать индивидуально, учитывая эффективность и токсичность препарата. Следует отметить, что при анализе отдаленных результатов исследования (через 24 месяца) было выявлено ухудшение показателей функции внешнего дыхания и рентгенологической картины, которые фактически возвращались к исходным значениям [80]. В рандомизированном двойном слепом исследовании была сопоставлена эффективность микофенолата мофетила (3000 мг/сут в течение 24 мес) и циклофосфамида внутрь (2 мг/кг/сут в течение 12 мес с дальнейшим приемом плацебо в течение 12 мес) у пациентов с ССД. Улучшение показателей функции внешнего дыхания было сопоставимым в двух группах, однако переносимость микофенолата мофетила была лучше, чем циклофосфамида [81].
В исследовании случай-контроль лечение ритуксимабом пациентов с ССД и интерстициальным поражением легких предотвращало снижение ФЖЕЛ по сравнению с контролем (p=0,02) [82]. Л.П. Ананьева и соавт. применяли ритуксимаб у 27 пациентов с ССД и поражением легких. У 82% пациентов был достигнут хороший эффект, а у 15% – удовлетворительный. Через 1 год после первого введения ритуксимаба было выявлено достоверное увеличение среднего значения ФЖЕЛ. Эффективность препарата у пациентов с длительностью болезни менее 5 лет была выше, чем у больных с более длительным анамнезом ССД: увеличение ФЖЕЛ на 11,4±12,2% и 3,7±5,7%, соответственно [83]. Мы применяли ритуксимаб у 8 пациентов с ССД и интерстициальным поражением легких. Через 6 мес ФЖЕЛ и диффузионная способность легких увеличились на 6,9±6,9% и 4,7±4,7% соответственно, а через 12 месяцев – на 8,9±6,9% и 6,7±4,6%.
В рандомизированном, двойном-слепом, плацебоконтролируемом исследовании лечение тоцилизумабом, блокирующим рецепторы интерлейкина-6, у 87 пациентов с ССД и интерстициальным поражением легких задерживало прогрессирование фиброза через 24 и 48 недель [84].
Склеродермический почечный криз. Основа лечения – ингибиторы АПФ, в частности каптоприл, который назначают в дозе 6,25-12,5 мг три раза в сутки, а затем постепенно увеличивают дозу до максимальной (50 мг три раза в сутки) [85]. Каптоприл не отменяют, даже если функция почек продолжает ухудшаться. После стабилизации АД можно перейти на прием ингибитора АПФ длительного действия. Постоянный прием ингибитора АПФ увеличил выживаемость пациентов со склеродермическим почечным кризом с 10% в течение года до 60% в течение 5 лет [86,87]. Если на фоне приема каптоприла в максимальной дозе не удается добиться нормализации АД в течение 72 ч, добавляют блокаторы кальциевых каналов, нитраты (особенно при появлении застойных явлений в легких) или другие вазодилатирующие средства. При сохранении олигурии может потребоваться лечение гемодиализом. Медлен ное (в течение 2 лет) восстановление или улучшение функции почек после склеродермического почечного криза происходит у 30% пациентов [88]. Если через 2 года сохраняется потребность в гемодиализе, возможна трансплантация почки [89].
Интересно отметить, что независимым фактором риска почечного склеродермического криза считают применение глюкокортикостероидов в дозе более 15 мг/сут, что недавно было показано в двух крупных исследованиях [90,91]. Возможно также развитие так называемого “нормотензивного” почечного криза, при котором АД не превышает 140/90 мм рт. ст. Выживаемость пациентов с нормотензивным кризом значительно ниже таковой пациентов с гипертензивным кризом – 13% и 35%, соответственно, ввиду поздней диагностики [92]. Рекомендаций по лечению данного состояния нет, в связи с чем терапия аналогична лечению гипертензивного почечного криза.
Поражение желудочно-кишечного тракта. При наличии гастроэзофагеального рефлюкса рекомендуется применение ингибиторов протонной помпы [93]. При необходимости могут быть использованы прокинетики [94]. Лечение антибактериальными препаратами для профилактики избыточного бактериального роста в тонкой кишке представляется спорным, учитывая отсутствие соответствующих рандомизированных клинических исследований [95].
Ранняя ССД. Количество исследований, в которых изучались подходы к лечению пациентов с ранней ССД, ограничено. В достаточно крупном проспективном обсервационном исследовании ESOS (European Scleroderma Observational Study) у 326 пациентов c диффузной формой ССД длительностью не более 3 лет изучалась эффективность различных режимов иммуносупрессивной терапии, в том числе метотрексатом (n=65), микофенолата мофетилом (n=118) и циклофосфамидом (n=87). Пятьдесят шесть пациентов не получали иммуносупрессивную терапию. 276 (84,7%) пациентов завершили 12-месячное наблюдение и 234 (71,7%) – 24-месячное. Во всех группах пациентов, получавших иммуносупрессивные препараты, отмечалось значительное снижение кожного счета по Rodnan. Через 24 месяца выживаемость была ниже в контрольной группе пациентов, хотя разница между группами не достигла статистической значимости [96].
Применение глюкокортикостероидов. Применение глюкокортикостероидов для лечения ССД по возможности следует ограничивать в связи с риском развития отдаленных осложнений, однако при прогрессирующем диффузном поражении кожи и других клинических признаках воспалительной активности (серозит, миозит, прогрессирующее интерстициальное поражение легких, рефрактерный синовит и/или теносиновит) целесообразно назначение глюкокортикостероидов в дозах до 15 мг/сут, так как прием этих препаратов в более высоких дозах увеличивает риск развития склеродермического почечного криза.
Заключение
ССД остается одним из самых неблагоприятных диффузных заболеваний соединительной ткани ввиду частого тяжелого поражения жизненно важных органов и ограниченных возможностей терапии. Многие вопросы лечения ССД остаются нерешенными, а прогнозирование эффективности терапии представляется затруднительным в связи с малым количеством исследований. Концепция ранней и очень ранней ССД, а также “окна возможностей” представляет интерес, однако опыт назначения иммуносупрессивного лечения в столь ранние сроки весьма ограничен. Перспективно дальнейшее изучение эффективности ритуксимаба, тоцилизумаба и, возможно, других ГИБП, однако до сих пор остается неясным, имеют ли препараты данной группы реальное преимущество перед стандартными препаратами.



