Агрегация тромбоцитов
Для оценки функции тромбоцитов в Лаборатории ЦИР проводится анализ на индуцированную агрегацию тромбоцитов. Это анализ высокого качества, выполняется на автоматическом агрегометре. Так как этот анализ резко меняется при приеме препаратов, влияющих на свертывание крови (антиагреганты, например, аспирин, тромбо-асс, антикоагулянты, например, гепарин), желательно сдавать его до начала приема этих лекарств. По каждой агрегатограмме врач-лаборант выдает заключение.
 Анализ на агрегацию тромбоцитов рекомендуется в следующих случаях: при
Анализ на агрегацию тромбоцитов рекомендуется в следующих случаях: при 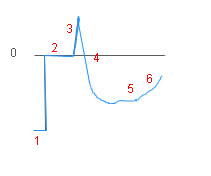 невынашивании беременности, неудачных попытках ЭКО, тяжелых осложнениях беременности в анамнезе, бесплодии неясного генеза, а также при повышенной кровоточивости: легкое образование синяков, меноррагии, носовые кровотечения.
невынашивании беременности, неудачных попытках ЭКО, тяжелых осложнениях беременности в анамнезе, бесплодии неясного генеза, а также при повышенной кровоточивости: легкое образование синяков, меноррагии, носовые кровотечения.
В кривой агрегации оцениваются амплитуда агрегации, форма кривой, наличие одной или двух волн, а также наличие дезагрегации.
Важная информация: сочетание приема пищевых продуктов, фитопрепаратов и пищевых добавок, содержащих компоненты из данного списка, с приемом антиагрегантов (тромбоАСС) и антикоагулянтов (гепарин) является опасной по риску кровотечения комбинацией (категория D по классификации FDA). Риск кровотечения в большинстве случаев превышает потенциальную пользу.
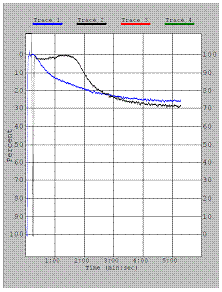 Агрегация с АДФ (синяя линия) и арахидоновой кислотой. Агрегационный ответ резко снижен. Дезагрегация практически отсутствует.
Агрегация с АДФ (синяя линия) и арахидоновой кислотой. Агрегационный ответ резко снижен. Дезагрегация практически отсутствует.
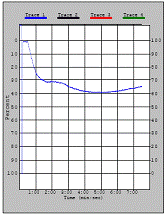
Агрегация с АДФ.
Агрегационный ответ снижен. Дезагрегация практически отсутствует.
Антиагреганты в практике лечения сердечно-сосудистых заболеваний
Тромбообразование играет ключевую роль в развитии различных сердечно-сосудистых осложнений. В патогенезе ишемических болезней органов и систем человеческого организма: ишемической болезни сердца (ИБС) [острый коронарный синдром (ОКС), инфаркт миокарда
Тромбообразование играет ключевую роль в развитии различных сердечно-сосудистых осложнений. В патогенезе ишемических болезней органов и систем человеческого организма: ишемической болезни сердца (ИБС) [острый коронарный синдром (ОКС), инфаркт миокарда (ИМ)], ишемического мозгового инсульта, гангрены конечностей и других нарушений кровоснабжения органов и тканей — значительное место занимают воспалительные и атеросклеротические повреждения сосудов с нарушением целостности интимы, замедление кровотока, дисбаланс свертывающей и противосвертывающей системы и нарушение реологических параметров крови.
При повреждении эндотелия сосудов различной этиологии происходит формирование тромба и вследствие этого — критическое сужение просвета сосудов или его полное закрытие (частичная или полная окклюзия). В нарушениях реологических параметров крови агрегационное состояние тромбоцитов и эритроцитов играет ведущую роль [1, 6, 8, 10, 17].
Современное лечение и профилактика сердечно-сосудистых осложнений, в частности ИМ, невозможны без четкого представления механизмов тромбообразования, материальным субстратом которого являются тромбоциты, эритроциты, фибриноген. ИМ — ишемический некроз сердечной мышцы — в 95% случаев развивается в результате формирования тромба в коронарной артерии, что подтверждается данными аутопсии среди умерших в первые 3 ч от начала развития болевого синдрома и результатами коронарографического исследования у больных, поступивших в стационар в первые часы развития миокардиальной катастрофы [10].
В процессе тромбообразования тромбоцит проходит четыре стадии: активация, высвобождение активных биологических веществ (тромбоксаны, аденозиндифосфат АДФ, серотонин, гликопротеиновые (ГП) рецепторы — IIb/IIIa), агрегация и адгезия. Эти процессы (макро- и микротромбообразование) особенно бурно проявляются при дестабилизации коронарного кровотока — ОКС,, включая ИМ. В основе макро- и микротромбообразования лежат механизмы, стимулирующие агрегационную активность тромбоцитов и эритроцитов — наличие ускоренного, турбулентного тока крови в суженном атеросклеротической бляшкой участке сосуда, что способствует повреждению эндотелия (с развитием эндотелиальной дисфункции) и «обнажению» коллагена, одного из главных факторов агрегации и адгезии тромбоцитов. Тромбоциты вступают в контакт с субэндотелиальным слоем, в частности с главным стимулятором адгезии — коллагеном, образуют отростки с образованием тромбоцитарных конгломератов (агрегация) и приклеиваются (адгезия) на этих участках, создавая белый тромб.
Активация тромбоцитов осуществляется катехоламинами, тромбином, АДФ, серотонином, коллагеном, тромбоксаном А2 — продуктом метаболизма арахидоновой кислоты.
В результате активации тромбоцитов происходит высвобождение из последних биологически активных веществ (АДФ, тромбоксан А2, серотонина) и лабилизация мембраны тромбоцита с образованием ГП-рецепторов IIb/IIIa под действием АДФ и тромбоксана А2.
Агрегация тромбоцитов — образование тромбоцитарных конгломератов в плазме крови — происходит при активации и взаимодействии ГП-рецепторов IIb/IIIa через образования фибриновых мостиков между тромбоцитами.
Адгезия — прилипание тромбоцитарных конгломератов к поврежденной интиме сосудов — контролируется фактором Виллебранда.
Таким образом, активация тромбоцитов — ключевой момент в патогенезе сердечно-сосудистых осложнений, во многом определяющий выраженность нарушений кровоснабжения органов и тканей (сердце, головной мозг, периферические сосуды), поэтому антиагрегационная терапия является патогенетически обоснованной.
В настоящее время выделяют четыре группы антитромбоцитарных препаратов, в основе разделения которых лежат принципы доказательной медицины, учитывающие эффективность и наличие побочных действий. Первая группа антитромбоцитарных препаратов, применение которых не рекомендуется для практической кардиологии вследствие отсутствия доказательной основы преимуществ перед Аспирином, неэффективности и потенциальной опасности: сульфинпиразон, дипиридамол, простациклин, блокаторы синтетазы тромбоксана А2, антагонисты рецепторов тромбоксана А2, препараты ингибиторов рецепторов IIb/IIIa тромбоцитов для приема внутрь. Вторая группа, составляющая основу современной антитромбоцитарной терапии, — ингибиторы циклооксигеназы (ЦОГ): ацетилсалициловая кислота (АСК) — Аспирин. Третья группа — тиенопиридины (клопидогрел — Плавикс, тиклопидин — Тиклин). Четвертая группа — блокаторы ГП-рецепторов IIb/IIIa для внутривенного применения (абциксимаб, эптифибатид, тирофибан, фрамон) [1, 3, 8, 9, 11, 12].
АСК используется в медицинской практике более ста лет. Начиная с 1980-х годов, Аспирин активно применяется в клинической практике при лечении ОКС с целью предупреждения развития ИМ. Проведено несколько значительных исследований (VA, RISC, ISIS-2), которые убедительно продемонстрировали способность Аспирина снизить риск развития острого ИМ и смерти от острой коронарной недостаточности на 41–70%. В 2002 г. Antithrombotic Trialists Collaboration опубликовала данные крупнейшего метаанализа результатов 287 рандомизированных клинических исследований, охвативших 135000 пациентов. Полученные данные свидетельствуют о преимуществах Аспирина у пациентов с высоким риском развития осложнений. В метаанализе убедительно показано, что применение высоких доз Аспирина (500–1500 мг) не имеет преимуществ в терапевтической эффективности по сравнению со средними (160–325 мг) и низкими (75–150 мг) дозами и довольно часто сопровождается побочными действиями со стороны желудочно-кишечного тракта (ЖКТ). Подобные результаты легли в основу постулата: оптимальная доза Аспирина для длительной профилактики сердечно-сосудистых осложнений у пациентов с высоким риском находится в пределах 75–150 мг/сут [1, 4, 8, 11, 13, 17].
Американская диабетологическая ассоциация рекомендует применять АСК в дозе от 81 до 325 мг для вторичной профилактики поражений крупных сосудов у больных диабетом старше 30 лет, сочетающегося с компонентами метаболического синдрома: избыточной массой тела (> 120% от идеальной массы тела, индекс массы тела (ИМТ) > 28 для женщин и > 27,3 для мужчин), атерогенной дислипидемией (триглицериды > 250 мг%, холестерин липопротеидов низкой плотности > 130 мг% и холестерин липопротеидов высокой плотности
А. М. Шилов, доктор медицинских наук, профессор
С. А. Князева, кандидат медицинских наук
С. А. Мацевич
ММА им. И. М. Сеченова, Москва
Филаргин обладает способностью вызывать агрегацию фибрил чего
В настоящее время имеются научные доказательства о генетически детерминированных нарушениях барьерных свойств кожи, что облегчает проникновение аллергенов в глубь кожи, повышает склонность к воздействию раздражающих факторов и, в конечном итоге, способствует воспалению. Дефицит филаггрина — наиболее изученная аномалия, в результате которой возрастает трансэпидермальная потеря воды (ТЭПВ). Помимо этого, дефицит межклеточных липидов в роговом слое и нарушенное соотношение между холестерином, незаменимыми жирными кислотами и церамидами усиливает ТЭПВ, что обусловливает образование эпидермальных микротрещин. Нарушение целостности барьерного слоя кожи ведет к нарушению метаболизма кожи и воспалению, что является ключевым промежуточным звеном патогенеза атопического дерматита (АтД) и ряда других дерматологических заболеваний. Базовым элементом терапии АтД, помимо устранения контакта со специфическими и неспецифическими провоцирующими факторами, является восстановление нарушенной барьерной функции кожи путем применения топических гидратирующих и защитных средств. Наружное применение смягчающих средств — одна из важных стратегий лечения АтД и многих других дерматозов для восстановления барьерной функции кожи.
Структура и свойства кожи
Кожа — самый большой орган человека; ее масса составляет 11—15% от массы тела. Кожа обеспечивает несколько важнейших функций: является барьером, отделяющим окружающую среду от внутренней; защищает от механических, тепловых, химических повреждений; регулирует количество воды в организме; обеспечивает осязание; защищает от инвазии патогенов, обеспечивает персистенцию симбиотических микроорганизмов; помогает вырабатывать витамин D и ряд гормонов.
Одним из наиболее важных белков, участвующих и регулирующих ороговение, является филаггрин. В процессе дифференцировки кератиноцитов в корнеоциты филаггрин формируется из предшественника белка — профилаггрина, который сохраняется в кератогиалиновых гранулах. Высвобождение и модификация профилаггрина в филаггрин вызывают агрегацию кератиновых филаментов и гибель клеток. Мутации в гене филаггрина часто выявляют у больных при АтД, астме и других дерматологических заболеваниях [3, 4].
Поверхностный слой эпидермиса состоит уже из частиц, которые постепенно отшелушиваются (см. рисунок, г). Для удержания чешуек вместе и сохранения целостности барьера кожа вырабатывает липидный клей, состоящий преимущественно из церамидов.
Церамиды и другие липиды рогового слоя
Структурно церамид состоит из двух молекул: сфингозидного полярного основания и жирной гидрофобной кислоты, соединенных амидной связью (см. рисунок, д).
Сфингозид через систему мембран аппарата Гольджи связан с клетками рогового слоя; жирные кислоты заполняют межклеточное пространство перпендикулярно пластам клеток (см. рисунок, е, ж). Красной линией отмечено расположение липидов между слоями клеток. Церамиды в области полярной части, прикрепленной к «мертвым» кератиноцитам, формируют малоподвижную псевдокристаллическую решетку; средняя часть липидной прослойки образована хвостами жирных кислот, имеющих меньший объем, чем сфингозиды, что обеспечивает их большую подвижность. Между ними пространство заполняется холестерином и СЖК, не связанными с церамидами, что обеспечивает текучесть (см. рисунок, е, ж) среднего пласта липидов. Таким образом, церамиды обеспечивают плотность рогового слоя, а средняя зона обеспечивает его эластичность.
Анализ структуры церамидов методом обращенно-фазовой жидкостной хроматографии в сочетании с квадрупольной времяпролетной масс-спектрометрией высокого разрешения показал, что вариантов комбинаций сфингозинов с жирными кислотами может быть более 1000 [6, 7]. В целом церамиды получаются комбинацией вариантов сфингозиновой полярной части и жирных кислот с разным числом атомов углерода (см. таблицу).  Строительные блоки церамидов рогового слоя кожи млекопитающих В настоящий момент используют буквенную номенклатуру церамидов, где сфингозин обозначается S, фитосфингозин — Р, 6-гидроксисфингозин — Н и дигидросфингозин — dS (см. таблицу). Жирные кислоты, выявленные в составе церамидов, также представлены четырьмя типами: кислоты, не содержащие гидроксил в α позиции (N), содержащие гидроксил в положении α или ω атома углерода (А и О соответственно) и этерифицированный гидроксил в положении ω (ЕО), что в сумме дает 16 классов церамидов. В каждом классе длина хвоста жирной кислоты может быть различной.
Строительные блоки церамидов рогового слоя кожи млекопитающих В настоящий момент используют буквенную номенклатуру церамидов, где сфингозин обозначается S, фитосфингозин — Р, 6-гидроксисфингозин — Н и дигидросфингозин — dS (см. таблицу). Жирные кислоты, выявленные в составе церамидов, также представлены четырьмя типами: кислоты, не содержащие гидроксил в α позиции (N), содержащие гидроксил в положении α или ω атома углерода (А и О соответственно) и этерифицированный гидроксил в положении ω (ЕО), что в сумме дает 16 классов церамидов. В каждом классе длина хвоста жирной кислоты может быть различной.
Синтез церамидов
В состав церамидов чаще всего входят длинноцепочечные насыщенные жирные кислоты, содержащие от 14 до 26 атомов углерода. Церамиды в организме образуются тремя различными путями, а именно синтезируются de novo в эндоплазматическом ретикулуме клеток из серина и пальмитата в результате гидролиза сфингомиелинидазой сфингомиелина, являющегося составной частью клеточных мембран, и из остаточного сфингозина [8]. Синтез церамидов прямо зависит от количества жировых отложений в организме. Так, уровень сывороточных церамидов С16:0, С18:0, С24:0 и С24:1 достоверно повышен у людей с ожирением [8].
Характеристика липидного состава при заболеваниях
Впервые церамиды были открыты в головном мозге, откуда и получили свое название (cerebrum). Церамиды также являются компонентами липопротеидов крови. Концентрация церамидов в мозге и крови значительно ниже, чем в коже. Изменение количества церамидов в крови часто имеет диагностическое значение, например при болезни Альцгеймера [8—11].
В норме соотношение церамидов, холестерина и СЖК составляет 3:1:1. С возрастом снижается продукция липидов кожи, но соотношение не меняется [12]. При различных метаболических нарушениях концентрация липидов кожи, а также церамидов крови может служить прогностическим и диагностическим маркерами сердечно-сосудистых заболеваний, ожирения, сахарного диабета, инсулинорезистентности и неалкогольной жировой болезни печени [13—16].
При АтД нарушение состава липидов кожи ассоциировано с расстройством синтеза как церамидов, так и СЖК. M. Danso и соавт. [17] показали, что при АтД снижаются количество насыщенных и доля длинноцепочечных (С22—С28) СЖК, а также изменяется баланс церамидов разных классов. Так, повышается доля AS и NS церамидов и снижается доля ЕОН и ОН церамидов. Изменения в составе СЖК и церамидов ассоциированы с нарушением функциональной активности ферментов стеарол CoA десатуразы (церамиды) и элонгазы 1 (СЖК). Аналогичное повышение доли AS и NS церамидов выявляют также у детей с АтД [18].
Нарушение в составе и количестве церамидов кожи наблюдается также при акне. В целом разнообразие церамидов в коже больных не нарушено. Так, Pappas и соавт. идентифицировали 283 типа церамидов при снижении общего количества липидов и доли NH, AH, EOS и EOH церамидов [19]. При акне (так же, как и при АтД) наблюдают снижение количества длинноцепочечных (>С18) СЖК. Авторы заключили, что NH и AH церамиды наиболее важны для формирования нормального барьера кожи [19].
Имеются ограниченные данные по изменению состава и количества церамидов при псориазе. Показано снижение церамида EOS [20]. В то же время состав церамидов на не пораженных псориазом и АтД участках кожи не отличается от такового у здоровых доноров [21]. Данных по составу и количеству церамидов в коже больных розацеа нет. По-видимому, при розацеа липиды кожи находятся в пределах нормы. В большинстве случаев нарушение состава липидов кожи ассоциировано с повышенной ТЭПВ. Имеются ограниченные сведения, что при розацеа ТЭПВ повышается только в области лица, что может являться в большей степени результатом патологического процесса, чем генерализованного дисбаланса липидов кожи [22]. Этими же авторами показано, что при АтД повышенная ТЭПВ является генерализованной.
Компенсаторные механизмы при нарушении барьерной функции эпидермиса
При нарушении эпидермального барьера (травмы, воспаление, аутоиммунные процессы) в течение минут начинаются репаративные процессы. В первую очередь высвобождаются ЛТ из клеточных депо и начинается синтез de novo СЖК, а затем церамидов [23]. При наложении непроницаемой мембраны наблюдается подавление синтеза ЛТ, что препятствует восстановлению эпидермального барьера [24]. Процесс репарации стимулируется изменением градиента кальция в эпидермисе, вызванного локальной потерей воды [25]. Нанесение на кожу топических препаратов, содержащих СЖК, ускоряет восстановление барьерных свойств кожи за счет включения экзогенных СЖК в липидный слой эпидермиса [26]. Аналогичное действие оказывают и синтетические церамиды [27]. Однако в состав топических средств входит несколько компонентов, создающих на коже пленку. В большей степени они служат окклюзивным барьером, помогающим удерживать воду и снижать ТЭПВ, уменьшать зуд и обеспечивать экзогенными липидами и церамидами [28].
Терапия кожи увлажняющими средствами
Показано, что использование увлажняющих препаратов замедляет прогрессию АтД и снижает тяжесть заболевания [30, 31]. K. Mori и соавт. [30] оценивали эффект геля на основе синтетических церамидов и экстракта эвкалипта в слепом клиническом исследовании 27 больных из Японии с умеренным АтД. Авторы показали, что в сухой летний период использование геля значительно улучшало состояние кожи, снижало покраснение, зуд, улучшало самочувствие больных по сравнению с больными, не использовавшими гель. Аналогичные данные были получены у больных себорейным дерматитом [31]. Этот же состав увлажняющего средства в сочетании с умеренной очисткой лица оказывал достоверный протективный эффект при акне средней тяжести у подростков с сухой и чувствительной кожей [32]. Авторы показали увеличение общего количества церамидов кожи, доли длинноцепочечных церамидов NS и NP в результате обработки кожи.
Использование топических средств, содержащих компоненты ЛТ, позволяет снизить побочные эффекты кортикостероидных препаратов. Так, короткий курс 0,05% клобетазола замедлял регенерацию эпидермального барьера; при одновременном нанесении крема, содержащего СЖК, холестерол и церамиды, снижались побочные эффекты кортикостероида и ускорялась репарация кожи [33]. Аналогичные данные были получены S. Ahn и соавт. [34] в модели на мышах.
Иммуносупрессивные препараты, влияющие на физиологические процессы кожи, подавляют репарацию: так, ингибиторы кальциневрина пимекролимус и такролимус задерживают восстановление барьерной функции и снижают количество липидов в эпидермисе [35]. Использование топических препаратов, включающих физиологические липиды в сочетании с пимекролимусом, улучшает репарацию кожи и состав липидов эпидермиса.
Применение в качестве наружной терапии воспалительных дерматозов комбинации топических кортикостероидов или ингибиторов кальциневрина с препаратами, содержащими физиологические липиды, позволяет улучшить восстановление эпидермального барьера и снизить побочные эффекты противовоспалительных агентов на барьерную функцию кожи.
В России разработан комбинированный препарат, содержащий 0,1% метилпреднизолона ацепонат и керамиды (Комфодерм К крем, патент 2011120522.15), который позволяет расширить возможности терапии стероидчувствительных дерматозов у взрослых и детей.
На рынке имеется большое количество кремов, содержащих церамиды. Чаще всего в косметические кремы вводятся церамиды NP и EOS, что способствует снижению ТЭПВ. В увлажняющие препараты и терапевтические кремы для лечения псориаза могут добавляться церамиды AP, AS и EOS. В настоящее время в основном используют синтетические церамиды, которые по действию идентичны природным. Несмотря на известное соотношение церамидов, холестерина и СЖК в нормальной коже, применение увлажняющих кремов, содержащих большое количество церамидов разных классов, может быть нецелесообразным. Так, значительный клинический эффект при умеренном АтД получен при использовании крема РС-104 на основе смеси амидов пальмитиновой кислоты, глицирретиновой кислоты и экстракта виноградных косточек [36]. Раннее начало использования эмолиентов с церамидами значительно снижает частоту заболеваемости АтД у младенцев из группы риска [37, 38]. Синтетические фитоцерамиды NP и EOP эффективны также при псориазе [39].
Заключение
Мутации гена филаггрина как фактор нарушения регуляции эпидермального барьера у детей
В статье представлены современные данные о роли мутаций гена филаггрина в формировании аллергических болезней.
The article presents current data of the role of filaggrina gene mutations in the formation of allergic diseases in children.
На сегодняшний день установлено, что мутации гена филаггрина (FLG) являются важными генетическими факторами риска развития атопического дерматита (АтД). Филаггрин — это один из ключевых белков, присутствующий в эпидермисе кожи уже на третьем месяце жизни ребенка и препятствующий потере воды через роговой слой [1, 2].
Ранее показано, что «атопический марш» предполагает тенденцию развития пищевой аллергии, бронхиальной астмы (БА) и аллергического ринита (АР) у больных с АтД. При этом мутации гена FLG являются факторами риска на каждом шаге в пролонгированном процессе: формирование АтД [3], аллергической сенсибилизации [4], БА у больных с АтД [5] и АР [6]. Таким образом, мутации гена FLG являются важным фактором риска реализации атопии в целом, но с разными шансами для конкретного фенотипа [7].
Тесная связь между мутациями гена FLG и АтД стала важной вехой в генетическом исследовании аллергических заболеваний [7]. Мутации гена FLG объясняют пониженную экспрессию FLG и продуктов его деградации у пациентов с АтД [2]. В настоящее время мутации гена FLG рассматривают как основной фактор риска развития АтД [8, 9], особенно у пациентов с ранним его началом (до 2 лет жизни) и больных с персистирующей БА [10].
В 2006 г. впервые обнаружены две нулевые мутации, р.R501X и с.2282del4, гена FLG у пациентов с вульгарным ихтиозом (наследственным заболеванием, связанным с нарушением кератинизации) в 15 семьях шотландского, ирландского и американцев европеоидного происхождения [3, 11]. При более детальном исследовании привлек внимание факт наличия у нескольких членов семей с ихтиозом также и АтД [12].
Впервые C. N. Palmer и соавт. [13] сообщили о снижении или отсутствии экспрессии гена FLG вследствие мутаций, связанных с потерей его функций, что ведет к нарушению эпидермального барьера и клиническим проявлениям АтД. На сегодняшний день идентифицировано около 40 мутаций гена FLG в европейских и азиатских популяциях [12–14].
Распространенность мутаций гена FLG различается по странам Европы, но р.R501X и с.2282del4 являются двумя наиболее часто встречающимися и имеющими существенную связь с АтД по всему континенту [15]. Однако в Италии данные мутации редки (частота аллелей менее 1%). Таким образом, в различных популяциях к АтД могут предрасполагать различные генетические факторы, что определяет необходимость дальнейшего детального изучения [3, 16].
Большинство исследований показали, что мутации гена FLG ассоциированы с АтД и БА или только с АтД [17–19]. В европейской популяции у носителей мутаций гена FLG (р.R501X и с.2282del4) шанс заболеть АтД практически в 5 раз выше (ОШ = 4,78, 95% ДИ [3,31–6,92]) в сравнении с контролем (ОШ = 1,99, 95% ДИ [1,72–2,31]) [4].
Последние исследования демонстрируют статистически значимую ассоциацию мутаций гена FLG с астмой (ОШ = 1,48, 95% ДИ [1,32–1,66]), но при этом БА сочеталась с АтД (ОШ = 1,11, 95% ДИ [0,88–1,41]) [15]. В японской когорте спектр мутаций гена FLG практически полностью отличается от европейцев и североамериканцев [7]. Только две мутации (р.R501X и р.E2422X) определялись как у европейского, так и у азиатского населения [20, 21].
Исследования, проведенные в РФ, в выборке пациентов с АтД выявили частоту делеции с.2282del4, равной 9,1% (Т. И. Саликова и соавт.). Кроме того, показано, что в Республике Башкортостан данная мутация выявлена у 12,3% больных АтД и лишь у 2,23% индивидов контрольной группы [22]. Ученые из ближнего зарубежья, изучая частоту мутации с.2282del4 у больных аллергодерматозами, обнаружили данную делецию у 9,9% пациентов [23].
В научной литературе также имеются данные об ассоциации мутации с.2282del4 гена FLG с АтД, АР и развитием БА у детей. Ряд авторов указывает на статистически значимую ассоциацию делеции с.2282del4 гена FLG с АР, причем как без АтД (ОШ = 1,78, 95% ДИ [1,16–2,73]), так и при его наличии (ОШ = 2,84, 95% ДИ [2,08–3,88]) [4, 24].
Ген FLG находится в хромосомной области 1q21 в составе эпидермального дифференцировочного комплекса (ЭДК), представляющего собой массивный кластер генов, охватывающий регион около 1,9 млн пар нуклеотидов геномной ДНК человека и принимающий участие в терминальной дифференцировке эпидермиса и образовании рогового слоя [7, 25]. В данном регионе идентифицировано около 45 генов [12]. Там же расположены гены, кодирующие белки, участвующие в формировании рогового конверта: прекурсоры лорикрина, инволюкрина и малых пролин-богатых белков. Другие гены ЭДК кодируют белки, связанные с синтезом профилаггрина и трихохиалина, а также семейство S100A кальцийсвязывающих белков [25]. Таким образом, ЭДК является группой структурно и эволюционно связанных генов, при тесном сотрудничестве которых протекает сложный механизм дифференцировки эпидермиса, нарушение которого ведет к развитию АтД [12].
Термин «филаггрин» (от англ. filament aggregating protein — филамент, агрегирующий белок) впервые введен в 1981 г. для описания класса структурных белков рогового слоя эпидермиса [1]. На ультраструктурном уровне выявлено, что потеря функции гена FLG связана с дезорганизацией нитей кератина, нарушением синтеза ламеллярных телец и архитектоники пластинчатого бислоя [5].
Филаггрин способствует образованию белково-липидного конверта ороговевших клеток, который заменяет плазматическую мембрану при дифференциации кератиноцитов, перекрестно связанных трансглутаминазой. При этом образуется барьер, предотвращающий потерю воды и минимизирующий проникновение аллергенов и микроорганизмов [3].
Ген профилаггрин/филаггрин состоит из трех экзонов и двух интронов. Большая часть белка ЭДК кодируется в третьем экзоне, который является самым большим (более 12 т. п.н.). Филаггрин образуется из профилаггрина и является ключевым белком, способствующим терминальной дифференцировке эпидермиса и образованию защитного барьера кожи [1, 7]. Профилаггрин представляет собой высокофосфорилированный, гистидин-богатый белок с молекулярной массой около 500 кДа [26]. Наибольшее его количество расположено не выше двух слоев рогового слоя. Профилаггрин «хранится» в виде неактивной формы в нерастворимых гранулах кератогиалина зернистого слоя эпидермиса. При увеличении уровня кальция происходит дегрануляция кератогиалиновых гранул [1, 5]. При этом профилаггрин подвергается дефосфорилированию с образованием 10–12 практически идентичных, с массой около 37 кДа, пептидов филаггрина [27], которые связывают кератиновые филаменты. Это приводит к «сжатию» клеток зернистого слоя эпидермиса в сплюснутые чешуйки. Такой цитоскелет вместе с прикрепленными белками и компонентами мембраны претерпевает пространственное перекрестное сцепление с формированием ороговевшей клеточной оболочки — наружного барьерного слоя кожи [26]. Филаггрин участвует в аггрегации кератиновых тонофиламентов, образуя между ними аморфный матрикс. К ним присоединяются белки, полисахариды, липиды, аминокислоты, высвобождающиеся при начинающемся здесь (под влиянием гидролитических ферментов кератиносом и лизосом) распаде ядер и органелл. В итоге образуется сложное по составу соединение кератогиалин [27]. Далее, с помощью различных протеаз, филаггрин подвергается дальнейшей деградации до свободных аминокислот и их производных [1]. Свободные аминокислоты катаболизируются до составных компонентов «натурального увлажняющего фактора», таких как молочная, пирролидонкарбоксильная и уроканиновая кислоты, а также мочевина. Совместно они способствуют гидратации эпидермиса, тем самым повышая барьерную функцию кожи [3]. Кроме того, продукты распада FLG вносят вклад в формирование определенной кислотности кожи. Так, трансуроканиновая и пирролидонкарбоновая кислоты помогают поддерживать определенный градиент рН эпидермиса. Об этом свидетельствует более высокое поверхностное рН эпидермиса при наличии мутаций гена FLG [9].
«Кислотная мантия» рогового слоя обладает противомикробным эффектом. Доказано, что продукты распада филаггрина оказывают тормозящее влияние на рост золотистого стафилококка [28]. Также он изменяет активность целого каскада сериновых протеаз, необходимых для скоординированной эпидермальной дифференцировки и образования рогового конверта [3].
Имеются данные о возможной роли продуктов распада филаггрина в защите от УФ-лучей. В результате фотоизомеризации трансцисуроканиновой кислоты возникают молекулы спектра действия 280–310 нм, находящиеся в диапазоне ультрафиолетовой радиации. Более того, in vitro доказано, что цисуроканиновая кислота обладает иммуномодулирующим действием в человеческих кератиноцитах и лейкоцитах [3].
Филаггрин экспрессируется в коже, переднем преддверье носа и слизистой оболочке рта, но не в респираторном эпителии дыхательных путей или эпителии пищевода [6, 29]. Несмотря на то, что мутации FLG не носят органоспецифического характера, тем не менее есть основания полагать, что они косвенно участвуют в развитии атопических заболеваний «отдаленных» органов. Ассоциация мутаций FLG с другими атопическими нарушениями, вероятно, происходит именно в результате чрескутанной сенсибилизации аллергенами и/или вторично, посредством системного, иммунологического механизма стимуляции через нарушенный кожный барьер [7, 29].
Более того, факт, что БА встречается только у индивидов-носителей мутаций гена FLG с АтД, подтверждает гипотезу о том, что БА является вторичной по отношению к аллергической сенсибилизации, произошедшей после нарушения эпидермального барьера. Мутации гена FLG, по всей видимости, играют роль в хронизации заболевания и IgE-сенсибилизации у пациентов с АтД [19].
Таким образом, предположительно дефицит FLG в коже может предрасполагать к БА у детей c АтД, увеличивая риск сенсибилизации к аэроаллергенам. Однако не следует полагать, что развитие БА является неизбежностью даже у гомозиготных носителей мутаций гена FLG, приводящих к изменению его функции. Кроме того, даже у детей, сенсибилизированных аэроаллергенами, данные мутации следует рассматривать как обязательную предпосылку для развития АтД или БА [29]. Несмотря на то, что до 30% пациентов европейских когорт с АтД имеют нулевые мутации гена FLG, маловероятно, что только это может объяснить увеличение трансэпидермальной воды, которое наблюдается почти у всех пациентов с АтД в активной стадии болезни [30]. Более того, около 40% индивидов-носителей мутации гена FLG вообще не страдают АтД (среди здоровых лиц Северной Европы частота носительства мутаций достигает 12%) [31]. В связи с этим возникает предположение, что только присутствие мутаций гена FLG недостаточно для реализации АтД и/или Th2-адаптивного иммунного ответа. Очевидно, что определенную роль играют и другие генетические дефекты, а также факторы окружающей среды. Вероятно, существуют и такие нарушения эпидермального барьера, которые вносят вклад и/или модулируют эпикутанную сенсибилизацию, что приводит к формированию атопического фенотипа [31].
Таким образом, механизмы, с помощью которых мутации гена FLG вызывают заболевания дыхательных путей, до конца еще не ясны. Чрескожная сенсибилизация и вторичные иммунологические эффекты индукции Th2-цитокинов в эпителии — это только гипотезы, которые требуют дальнейшего детального исследования [7, 19].
Литература
С. В. Левашева 1
Э. И. Эткина, доктор медицинских наук, профессор
Л. Л. Гурьева, кандидат медицинских наук
Л. И. Бабенкова, кандидат медицинских наук
А. Р. Бикташева, кандидат медицинских наук
Н. А. Орлова, кандидат медицинских наук
А. А. Фазылова, кандидат медицинских наук
Г. Д. Сакаева, кандидат медицинских наук
Л. Я. Данилова
С. Э. Якута
ГБОУ ВПО БГМУ МЗ РФ, Уфа



