Ферментопатия что это такое
Синонимы синдрома ферментопатии. Химические пороки развития. Молекулярные болезни. Болезни ферментативной недостаточности. Врожденные аномалии обмена. «Врожденные ошибки обмена» (Garrod).
Определение синдрома ферментопатии. Групповое обозначение различных заболеваний, вызываемых (врожденным) отсутствием, недостаточностью или функциональной бездеятельностью (блокадой) определенного фермента или ферментативной системы. Встречаются как гетерозиготные, так и гомозиготные носители ферментативного дефекта. У последних ферментативная блокада является полной, у первых снижение соответствующей ферментативной активности достигает примерно половины нормальных показателей. В подобных случаях можно допустить, что дефект того или иного фермента не сопровождается какими-либо клиническими проявлениями в связи с компенсаторной активностью сохранившихся ферментов.
В патогенетическом отношении различают следующие типы ферментопатий в зависимости от характера ферментативного дефекта и его влияния на организм:
Тип I. Клинические признаки являются прямым следствием недостаточности или отсутствия биологически важного фермента.
Тип II. В результате ферментативной блокады скапливается физиологический субстрат, его концентрация в крови и (вторично) в моче резко увеличивается. Это вещество не оказывает повреждающего клетки действия, но его высокая концентрация в моче приводит к образованию камней.
Тип III. Физиологический субстрат, застаивающийся в результате ферментативных расстройств, в нефизиолсгических концентрациях действует токсически (а) или нарушает клеточный обмен веществ (б) в результате его отложений в клетках.
Тип IV. Многие вещества, состоящие в основной цепи, образуют в боковых цепях следы других продуктов расщепления, которые не оказывают влияния на обменные процессы. При блокаде основной цепи эти вещества меняются качественно и количественно и приобретают токсические свойства. Это наблюдается в тех случаях, когда отсутствует нормальный ферментативный механизм, который благодаря быстрому устранению побочных продуктов предотвращает их патологическое действие. Развивающаяся таким путем вторичная ферментопатическая недостаточность также может быть относительной, в частности, в тех случаях, когда первичный дефект основной ферментативной цепи способен захватывать избыточные количества побочных продуктов.
Тип V. Субстрат, скапливающийся в результате ферментативной блокады, образует токсические побочные продукты. Речь может идти лишь о таких веществах, для потенциального развития которых в клетке требуется необходимое воздействие ферментов.
Основные синдромы ферментопатий, известные в настоящее время
Тип I:
1. S. McArdle
2. Альбинизм
3. S. Rathbun
4. Анемия сидероахрестическая
5. S. Cooley и S. Rietti —Greppi—Micheli
6. Анемии ферментопенические гемолитические (первичного типа)
7. Метгемоглобинемия ферментопатическая
8. Оратацидурия
9. Идиопатические формы S. Dressier
10. S. Pendred
Тип II:
1. Оксалоз
2. Ксантинурия
3. Пентозурия
Тип III:
а) 1. S. Foiling
2. S. Ahornsirup
3. Галактоземия
4. Непереносимость фруктозы
5. S. Takahara
б) 1. S.v. Gierke:
— Форма I (печеночно-почечная форма)
— Форма II (S. Pompe)
— Форма III
— Форма IV
2. S. Crigler—Najjar
3. Недостаточность аргинин-янтарной кислоты
4. Цистатионурия
Тип IV:
1. Алкаптонурия
2. Адрено-генитальный синдром
Тип V:
1. S. Hartnup
2. Синдромы порфирии (S. Gunther I, синдром острой порфирии и синдром хронической порфирии)
Формы ферментопатий, не поддающиеся точной классификации и локализации
1. S. Wilson.
2. S. Troisier — Hanoi—Chauffard (гемохроматоз).
3. S. Abderhalden—Fanconi.
4. Синдром цистинурии.
5. Синдром несахарного диабета.
6. Почечный сахарный диабет.
7. Различные синдромы меллитурии.
8. Синдромы дефектопротеинемии.
9. Синдромы гемоглобинопатии.
10. Синдром недостаточности сахарозы.
11. Синдром врожденн ой недостаточности лактозы.
12. Синдром недостаточности пиридоксина.
13. Многочисленные болезни отложений и синдромы липидоза.
14. S. Lowe.
15. S. Gilbert—Lereboullet.
16. S. Greenfield. 17. S. Bernard.
18. Синдром амавротической идиотии.
19. S. Bessman—Baldwin.
20. Синдромы ферментопатического слабоумия.
21. S. Refsum.
22. S. Riley—Day.
23. S. Rowley—Rosenberg.
24. S. Silvestroni—Bianco.
25. S. Stein—Leventhal.
26. Синдромы тромбопатии.
27. S. Alexander.
28. Синдром анальбуминемии.
29. S. Bloom.
30. Синдром цитруллинурии.
31. Синдромы непереносимости дисахарида.
32. S. Glanzmann.
33. Синдром глюкоглицинурии.
34. Синдром глицинурии.
35. Синдром гликокола.
36. Синдром фавизма.
37. Синдром гистидинемии.
38. Синдром гомоцистинурии.
39. S. Hooft.
40. Синдром гидроксипролинемии.
41. Синдром гипераммонемии и др.
Этиология и патогенез. В большинстве случаев аутосомные рецессивно-наследственные расстройства с вариабельной пенетрантностью. Врожденные ферментативные дефекты развиваются благодаря тому, что нарушения генетической информации, связанные с микромолекулой ДНК хромосом, переносятся на РНК микросом. Последние влияют на нормальную и патологически измененную генетическую информацию в то время, как образуется фермент, способствующий специфической проводимости в клетки.

Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Ферментопатии
Полезное
Смотреть что такое «Ферментопатии» в других словарях:
ФЕРМЕНТОПАТИИ — то же, что энзимопатии … Большой Энциклопедический словарь
ферментопатии — то же, что энзимопатии. * * * ФЕРМЕНТОПАТИИ ФЕРМЕНТОПАТИИ, то же, что энзимопатии (см. ЭНЗИМОПАТИИ) … Энциклопедический словарь
Ферментопатии — Ферментопатия (лат. fermentopathia; фермент + греч. лат. pathos страдание, болезнь; син. энзимопатия) общее название болезней или патологических состояний, развивающихся вследствие отсутствия или нарушения активности каких либо ферментов. Смысл… … Википедия
Ферментопатии — (лат. fermentare – вызывать брожение греч. pathos – страдание, болезнь) – класс наследственных заболеваний, вызванных отсутствием или нарушением активности определённых ферментов, что влечёт серьёзные нарушения обмена веществ. Многие… … Энциклопедический словарь по психологии и педагогике
Ферментопатии — энзимопатии, заболевания, обусловленные врождённым дефектом обмена веществ вследствие ферментных нарушений; относятся к группе наследственных заболеваний (См. Наследственные заболевания). В основе Ф. лежат различные виды нарушений (полное … Большая советская энциклопедия
ФЕРМЕНТОПАТИИ — то же, что энзимопатии … Естествознание. Энциклопедический словарь
Наследственные болезни — I Наследственные болезни заболевания человека, обусловленные хромосомными и генными мутациями. Нередко ошибочно термины «наследственная болезнь» и «врожденная болезнь» употребляются как синонимы, однако врожденными болезнями называют те… … Медицинская энциклопедия
Первичные иммунодефициты — наследственные или приобретённые во внутриутробном периоде иммунодефицитные состояния. Обычно они проявляются или сразу после рождения, или в течение первых двух лет жизни (врождённые иммунодефициты). Однако менее выраженные генетические дефекты… … Википедия
Дистрофия клеток и тканей — нарушение тканевого или клеточного обмена, сопровождающееся определенными структурными изменениями клеток и межклеточного вещества. В основе развития дистрофии лежат расстройства регуляторных механизмов трофики врожденного или приобретенного… … Медицинская энциклопедия
Ферментная недостаточность поджелудочной железы
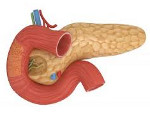
Ферментная недостаточность поджелудочной железы – это ограниченная секреция либо низкая активность панкреатических ферментов, приводящая к нарушению расщепления и всасывания питательных веществ в кишечнике. Проявляется прогрессивным похудением, метеоризмом, анемией, стеатореей, полифекалией, диареей и полигиповитаминозом. Диагностика основана на лабораторных методах исследования внешней секреции поджелудочной железы, проведении копрограммы, определении уровня ферментов в кале. Лечение включает терапию основного заболевания, нормализацию поступления нутриентов в организм, заместительное введение ферментов поджелудочной железы, симптоматическое лечение.
МКБ-10
Общие сведения
Ферментная недостаточность поджелудочной железы – одна из разновидностей пищевой интолерантности, которая развивается на фоне угнетения внешнесекреторной панкреатической деятельности. Оценить частоту экзокринной недостаточности ПЖ в популяции не представляется возможным, так как исследования, посвященные этому состоянию, практически не проводятся, а частота выявления ферментной недостаточности гораздо выше, чем, например, хронических панкреатитов.
Тем не менее, недостаточность выработки панкреатических ферментов является серьезным состоянием, способным привести к выраженному истощению и даже смерти пациента при отсутствии адекватного лечения. Практические изыскания в области гастроэнтерологии направлены на разработку современных ферментных препаратов, способных полностью заместить экзокринную функцию поджелудочной железы и обеспечить нормальное течение процессов пищеварения.
Причины
Первичная панкреатическая недостаточность связана с поражением поджелудочной железы и угнетением ее экзокринной функции. К причинам формирования первичной панкреатической недостаточности относят:
При вторичной форме патологии ферменты вырабатываются в достаточном количестве, однако в тонком кишечнике инактивируются либо их активация не происходит. Вторичная ферментная недостаточность поджелудочной железы развивается при:
Патогенез
Классификация
Недостаточность экзокринной функции поджелудочной железы может быть:
Абсолютная ферментная недостаточность поджелудочной железы обусловлена угнетением секреции ферментов и бикарбонатов на фоне уменьшения объема паренхимы органа. Относительная недостаточность связана со снижением поступления панкреатического сока в кишечник из-за обтурации просвета протоков поджелудочной железы камнем, опухолью, рубцами.
Симптомы ферментной недостаточности
В клинической картине ферментной недостаточности поджелудочной железы наибольшее значение имеет синдром мальдигестии (угнетение пищеварения в просвете кишечника). Непереваренные жиры, попадая в просвет толстого кишечника, стимулируют секрецию колоноцитов – формируется полифекалия и диарея (стул жидкий, увеличен в объеме), кал имеет зловонный запах, цвет серый, поверхность маслянистая, блестящая. В стуле могут быть видны непереваренные комочки пищи.
Мальдигестия протеинов приводит к развитию белково-энергетической недостаточности, проявляющейся прогрессирующим похудением, дегидратацией, дефицитом витаминов и микроэлементов, анемией. На продолжающуюся потерю веса большое влияние оказывает соблюдение диеты с ограничением жиров и углеводов, а также боязнь приема пищи, формирующаяся у многих пациентов с хроническим панкреатитом.
Нарушения моторики желудка (тошнота, рвота, изжога, чувство переполнения желудка) могут быть связаны как с обострением панкреатита, так и с опосредованным влиянием экзокринной панкреатической недостаточности за счет нарушения гастро-интестинальной регуляции, развития дуодено-гастрального рефлюкса и др.
Диагностика
Основное значение для выявления ферментной недостаточности поджелудочной железы имеют специальные тесты (зондовые и беззондовые), часто комбинирующиеся с ультразвуковыми, рентгенологическими и эндоскопическими методами. Зондовые методики являются более дорогостоящими и причиняют пациентам дискомфорт, однако и результаты их более точные. Беззондовые тесты дешевле, спокойнее переносятся больными, но они дают возможность определить панкреатическую недостаточность только при значительном снижении или полном отсутствии ферментов:
Кроме того, определить уровень панкреатической секреции можно и косвенными методами: по степени поглощения плазменных аминокислот поджелудочной железой, путем качественного анализа копрограммы (будет повышено содержание нейтральных жиров и мыла на фоне нормального уровня жирных кислот), количественного определения в кале жира, фекального химотрипсина и трипсина, эластазы-1.
Инструментальные методы диагностики (рентгенография органов брюшной полости, МРТ, КТ, УЗИ поджелудочной железы и гепатобилиарной системы, ЭРХПГ) используются для выявления основного и сопутствующих заболеваний.
Лечение ферментной недостаточности поджелудочной железы
Лечение экзокринной панкреатической недостаточности должно быть комплексным, включать коррекцию нутритивного статуса, этиотропную и заместительную терапию, симптоматическое лечение. Этиотропная терапия направлена, в основном, на предотвращение прогрессирования гибели паренхимы ПЖ.
Диетотерапия
Коррекция пищевого поведения заключается в исключении употребления алкоголя и табакокурения, увеличении количества белка в рационе до 150г/сут., сокращении количества жиров как минимум вдвое от физиологической нормы, приеме витаминов в лечебных дозировках. При выраженном истощении может потребоваться частичное либо полное парентеральное питание.
Медикаментозная терапия
Основным методом лечения ферментной недостаточности поджелудочной железы является пожизненный заместительный прием ферментов с пищей. Показания к заместительной ферментной терапии при панкреатической недостаточности: стеаторея с потерей более 15 г жира в стуки, прогрессирующая белково-энергетическая недостаточность.
Наибольшей эффективностью на сегодняшний день обладают микрогранулированные ферментные препараты в кислотоустойчивой оболочке, заключенные в желатиновую капсулу – капсула растворяется в желудке, создавая условия для равномерного перемешивания гранул препарата с пищей. В ДПК, при достижении уровня рН 5,5, содержимое гранул высвобождается, обеспечивая достаточный уровень панкреатических ферментов в дуоденальном соке. Дозировки препаратов подбираются индивидуально, в зависимости от тяжести заболевания, уровня панкреатической секреции. Критериями эффективности заместительной терапии и адекватности дозировок ферментных препаратов является увеличение веса, уменьшение метеоризма, нормализация стула.
Прогноз и профилактика
Прогноз при панкреатической недостаточности обусловлен выраженностью основного заболевания и степенью поражения паренхимы поджелудочной железы. Учитывая тот факт, что ферментная недостаточность поджелудочной железы развивается при гибели значительной части органа, прогноз обычно сомнительный. Предупредить развитие данного состояния можно путем своевременной диагностики и лечения заболеваний поджелудочной железы, отказа от приема алкоголя, курения.
Ферментная недостаточность у детей
Синдром мальабсорбции
— это синдром нарушения всасывания компонентов пищи в тонком кишечнике. Абсорбции или всасыванию мономеров (жирных кислот, аминокислот, моносахаров и др.) предшествует гидролиз — расщепление пищевых полимеров (белков, жиров и углеводов) под воздействием пищеварительных ферментов. Нарушение гидролиза полимеров при недостаточности пищеварительных ферментов (дигестивных энзимов) называется синдромом мальдигестии или синдромом недостаточности пищеварения. Часто встречающееся в клинической практике сочетание обоих видов расстройств — мальабсорбции и мальдигестии — предлагалось обозначать как синдром мальассимиляции. Однако в медицинской литературе традиционно называют мальабсорбцией все нарушения кишечного всасывания, вызванные как собственно патологией процесса абсорбции, так и недостаточностью пищеварительных ферментов. Синдромом мальабсорбции сопровождается целая группа заболеваний, в основе развития которых лежит наследственно обусловленная или вторичная (при патологии поджелудочной железы или других органов системы пищеварения) недостаточность ферментов.
На первом году жизни чаще всего синдром малъабсорбции является генетической наследственной патологией, наиболее известными наследственными ферментопатиями являются дисахаридазная недостаточность, целиакия и муковисцидоз.
Дисахаридазная недостаточность.
Дисахариды — компоненты большинства углеводов. Процессы их переваривания обеспечиваются специальными ферментами кишечника — дисахаридазами. После распада дисахаридов образуются моносахара, которые могут затем всасываться при помощи транспортных систем кишечника. Непереносимость дисахаридов у детей обусловлена наследственным отсутствием или снижением активности одной или нескольких дисахаридаз кишечника, в результате чего происходит неполное расщепление дисахаридов в тонкой кишке. Перистальтическими движениями кишечника не полностью расщепленные дисахариды перемещаются в нижние отделы пищеварительного тракта, где под действием естественной микрофлоры переходят в органические кислоты, сахара и водород. Эти вещества снижают абсорбцию воды и солей из полости кишечника, т. е. пищевая кашица (химус) разжижается, и это приводит к развитию поноса у ребенка. Симптомы первичной дисахаридазной недостаточности появляются у ребенка обычно сразу же после рождения. Для этой группы болезней характерно то, что с возрастом происходит некоторая компенсация нарушенных ферментативных функций, и симптомы болезни смягчаются или вообще проходят. Среди наследственных дефектов дисахаридаз наиболее известными являются недостаточность лактозы, сахарозы, изомальтазы, трегалазы. Недостаточность лактазы объясняется мутацией гена, который отвечает за синтез лактозы, в результате чего этот фермент или не синтезируется совсем (алактазия), или синтезируется его малоактивная форма (гиполактазия). Поэтому при поступлении лактозы в кишечник она полностью не расщепляется неполноценной лактозой, и развиваются характерный симптом мальабсорбции — диарея.
Родителям необходимо знать о том, что лактоза содержится не только в цельном женском, коровьем и козьем молоке, но и во всех видах сухого молока, во многих кисломолочных продуктах (сметане), сгущенном молоке, а также в некоторых медикаментах в качестве наполнителя. Поэтому при назначении лекарства следует информировать врача об имеющемся дефиците лактозы и внимательно ознакомиться с аннотацией препарата. В легких случаях непереносимости лактозы вместо молока дают молочные продукты и молоко, обработанное препаратами B-галактозидазы. Детям с лактозной недостаточностью показаны продукты, содержащие фруктозу (овощные и фруктовые пюре), которая хорошо всасывается и не подвергается бактериальному брожению. Кроме лечебного питания, в первые дни болезни коротким курсом (5—7 дней) назначают ферментативные препараты. В течение 30—45 дней применяют пробиотики для нормализации кишечной. Недостаточность сахарозы и изомальтазы чаще встречается вместе. У детей, находящихся на искусственном вскармливании, признаки дефицита этих ферментов отсутствуют. Симптомы заболевания появляются после употребления ребенком пищи, содержащей сахарозу и крахмал (сахар, картофель, манная крупа, мучные изделия) при переводе его на искусственное вскармливание или после введения прикормов. У ребенка после приема такой пищи возникают пенистый водянистый стул, рвота. При тяжелой форме и нахождении ребенка на искусственном вскармливании смесями, содержащими сахарозу, рвота становится упорной, ребенок теряет в весе. Диагноз непереносимости сахарозы подтверждают проведением пробы с нагрузкой сахарозой. Лечение состоит в соблюдении диеты с исключением продуктов, содержащих сахарозу и крахмал. Можно употреблять фрукты и овощи, в которых количество сахарозы невелико (морковь, яблоки). Прогноз благоприятный. С возрастом недостаточность ферментов компенсируется, и диету можно расширить.
Глютеновая болезнь (целиакия)
— это хроническое наследственное заболевание, развивающееся вследствие недостаточности ферментов, участвующих в переваривании глютена. В последние годы в связи с повышением качества диагностики глютеновая болезнь обнаруживается все чаще. Глютен — это компонент клейковины ряда злаковых культур — пшеницы, ржи, ячменя, овса. При распаде глютена образуется токсичный продукт — глиадин, который оказывает повреждающее действие на слизистую оболочку тонкого кишечника. Однако в норме у здоровых детей глиадин не повреждает слизистой, так как специфические ферменты расщепляют его до нетоксичных субстанций. При глютеновой болезни наблюдается выраженный в различной степени дефицит этих ферментов (вплоть до их полного отсутствия). В результате глютен не гидролизуется в кишечнике, а накапливается вместе с продуктами своего неполного расщепления, оказывая токсическое воздействие на слизистую оболочку тонкого кишечника, клетки слизистой тонкого кишечника погибают, и нарушается переваривающая и всасывательная функции. В типичных случаях глютеновой болезни заболевание имеет хроническое течение с периодами обострения и ремиссии.
Первые признаки целиакии появляются у ребенка во втором полугодии жизни после введения прикормов, в состав которых входит глютен злаков (манной, пшеничной, овсяной каш).
Если ребенок находится на искусственном вскармливании смесями, содержащими пшеничную, муку, то симптомы болезни проявляются раньше. От момента введения в питание ребенка глютенсодержащих продуктов до появления симптоматики проходит обычно 4—8 недель. Основными признаками глютеновой болезни являются дистрофия потеря в весе и отставание в росте, диарея, стеаторея (наличие нерасщепленных жиров в кале) и поражение центральной нервной системы. Клиника целиакии развивается постепенно. Сначала у ребенка снижается аппетит, появляются вялость, слабость, частые срыгивания. В дальнейшем срыгивания переходят в рвоту, развивается диарея. Кал при целиакии резко зловонный, обильный, пенистый, бледный с сероватым оттенком, блестящий. Ребенок перестает прибавлять в весе, а затем масса тела у него снижается. Дети сильно отстают в росте. Живот увеличен, что в сочетании с тонкими конечностями придает ребенку характерный внешний вид — «рюкзак на ножках». Выражение лица ребенка грустное, мимика скудная («несчастный вид»). Со временем поражаются и другие органы системы пищеварения — печень, поджелудочная железа, двенадцатиперстная кишка. Может развиваться цирроз печени. Отмечается умеренное увеличение в размерах печени и селезенки. Нарушение ферментообразующей функции поджелудочной железы приводит к еще большему угнетению процесса пищеварения. Возможно развитие вторичной недостаточности инсулина, чем объясняются симптомы сахарного диабета (повышенное выделение мочи и жажда) в период обострения болезни. Страдают все виды обмена веществ, особенно белкового. Развивается дефицит аминокислот, снижается концентрация общих липидов, холестерина и увеличивается количество кетоновых тел в сыворотке крови. Обменные нарушения проявляются рахитом, полигиповитаминозами, анемией. У детей может возникать облысение волосистой части головы (алопеция), часты переломы трубчатых костей. Развивается вторичное иммунодефицитное состояние, дети подвержены частым простудным заболеваниям, которые протекают более тяжело и длительно. У всех детей наблюдаются расстройства центральной нервной системы (обменно-токсическая энцефалопатия), дети раздражительны, капризны, отстают в психомоторном развитии. Для подтверждения диагноза целиакии применяют провокационный тест с глютеном и исследование биоптата слизистой оболочки двенадцатиперстной или тощей кишки.
Основным методом лечения глютеновой болезни является диетотерапия — исключение из питания больного ребенка всех продуктов, содержащих глютен (хлеба, хлебобулочных, кондитерских и макаронных изделий, манной, овсяной, перловой, пшеничной и ячневой круп и каш промышленного производства из этих круп). Так как мука и другие продукты переработки глютенсодержащих злаковых культур нередко добавляются в состав колбас, сосисок, сарделек, мясных и рыбных консервов (в том числе и предназначенных для детского питания), их тоже исключают из рациона ребенка. В острый период заболевания используют также ферменты, витамины, пробиотики.
При крайне тяжелых формах целиакии и низкой эффективности диетотерапии применяют глюкокортикоиды (гормоны) коротким курсом с постепенной отменой в последующем. Родители должны помнить о том, что безглютеновую диету соблюдать следует пожизненно, даже при полном исчезновении признаков заболевания. Несоблюдение диеты может привести к более тяжелому рецидиву заболевания, и возникшие нарушения тяжело будет компенсировать.
Кишечная форма муковисцидоза.
Генетический дефект при этом наследственном заболевании нарушает обратное всасывание хлорида натрия всеми экзокринными железами, в результате чего их секрет становится вязким, густым, отток его затруднен. Секрет застаивается в выводных протоках желез, они расширяются, и образуются кисты. Происходит гибель железистых клеток. Чаще всего встречаются кишечная и легочная форма муковисцидоза, при которых преимущественно поражаются соответственно железы кишечника или бронхов. Могут встречаться комбинированные формы. Поражение желез кишечника при муковисцидозе приводит к тому, что значительно нарушаются процессы переваривания и всасывания компонентов пищи (синдромы мальдигестии и мальабсорбции). В большей степени страдает переваривание жиров, что связано с угнетением ферментативной активности поджелудочной железы, которая тоже страдает при муковисцидозе.
У новорожденных кишечная форма муковисцидоза может проявиться мекониальным илеусом — кишечной непроходимостью в результате закупорки просвета кишечника густым и вязкиммеконием. В норме у новорожденных меконий должен отходить в первые сутки жизни. Если этого не происходит, должна возникать настороженность в отношении муковисцидоза, тем более, если у родителей или близких родственников ребенка имеются признаки этого заболевания. Осложнениями мекониального илеуса являются перфорация (образование отверстия) стенки кишечника и развитие тяжелого состояния — мекониального перитонита (воспаление брюшины).
На первом году жизни у ребенка с муковисцидозом, несмотря на хороший уход, рациональное вскармливание и хороший аппетит, плохо нарастает масса тела. Особенно ярко признаки муковисцидоза проявляются при переходе от грудного на смешанное или искусственное вскармливание. Характерен внешний вид детей, больных муковисцидозом: «кукольное» лицо, деформированная грудная клетка, большой вздутый живот, часто — пупочная грыжа. Конечности худые, отмечается деформация (утолщение) концевых фаланг пальцев в виде барабанных палочек. Кожа сухая, ее цвет серовато-землистый. Стул ребенка обильный, жидкий, сероватого цвета, со зловонным специфическим запахом прогорклого жира («мышиный запах»), жирный и блестящий, плохо смывается с горшка и пеленок. Часто встречается симптом «проскальзывания», когда стул у ребенка возникает сразу же после кормления. В некоторых случаях могут быть запоры, замазкообразная консистенция стула или выходу разжиженного кала предшествует выход каловой пробки. На фоне запоров часто возникает выпадение слизистой оболочки прямой кишки (как правило, хирургической коррекции в этом случае не требуется). Иногда не переваренный жир вытекает из заднего прохода ребенка в виде маслянистой жидкости, оставляя на пеленках жирный след. Если дефекация и мочеиспускание происходят одновременно, на поверхности мочи плавает жир в виде маслянистых пленок. Вследствие постоянного длительного нарушения процессов пищеварения ребенок на фоне обычно хорошего или повышенного аппетита худеет (вплоть до развития тяжелых форм истощения), появляются признаки недостаточности различных витаминов (полигиповитаминоз) страдают обменные процессы. Дети отстают в физическом развитии. Диагностика муковисцидоза проводится при помощи исследования кала (определение непереваренных остатков пищи), определения трипсина в кале, потовых проб (в потовой жидкости определяют содержание натрия и хлора) и при помощи специфического генетического исследования с идентификацией мутантного гена. Лечение при кишечной форме муковисцидоза направлено на улучшение процессов пищеварения. При тяжелом и средне-тяжелом состоянии ребёнка, когда синдром мальабсорбции сочетается с обезвоживанием и выраженным токсикозом, диету начинают с водно-чайной паузы. Ребенка отпаивают из расчета 100—150 мл/кг массы в сутки 5%-ным раствором глюкозы, регидроном, зеленым чаем и т. д. При тяжелом состоянии глюкозо-солевые растворы вводят также внутривенно капельно. В стационаре в острый период назначают гормональную терапию коротким курсом, витамины. После снятия обострения ребенок переводится на диету с частотой кормления 8—10 раз в сутки. Дети первого года жизни продолжают получать грудное молоко, которое является для них оптимальным видом пищи. При искусственном вскармливании предпочтение должно отдаваться смесям, которые содержат жиры в виде среднецепочечных триглицеридов. Эти компоненты жиров не требуют для своего переваривания участия ферментов поджелудочной железы и поэтому легко всасываются. После введения прикормов и в последующей жизни диета ребенка должна включать минимальные количества жиров и большие количества белка. Больные муковисцидозом нуждаются в повышенном количестве белка, так как он теряется в значительном объеме из-за синдрома мальабсорбции. Поэтому в питание ребенка с 7-месячного возраста необходимо включать такие высокобелковые продукты, как мясо, рыбу, яйцо и творог. Кроме диеты назначаются индивидуально подобранные дозировки ферментных препаратов. Для нормализации кишечного биоценоза применяют препараты нормальной кишечной флоры курсами по 2—3 месяца и более. Проводятся курсы витаминотерапии. Родителям следует внимательно следить за состоянием ребенка, а если в семье имеются указания на возможное наличие заболевания у родственников (сами родители могут быть и здоровы) — информировать об этом педиатра.



