Фенилкетонурия у детей и ее лечение
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотип
.jpg) Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Определение, патогенез, классификация
Фенилкетонурия — наследственная аминоацидопатия, связанная с нарушением метаболизма фенилаланина, в результате мутационной блокады ферментов приводящая к стойкой хронической интоксикации и поражению ЦНС c выраженным снижением интеллекта и неврологическим дефицитом [1, 2].
Основное значение в патогенезе классической ФКУ имеет неспособность фенилаланингидроксилазы перерабатывать фенилаланин до тирозина. В результате в организме накапливается фенилаланин и продукты его аномального обмена (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты) [1–3].
В числе других патогенетических факторов рассматриваются нарушения аминокислотного транспорта через гематоэнцефалический барьер, нарушения церебрального пула аминокислот с последующим нарушением синтеза протеолипидных белков, нарушения миелинизации, низкие уровни нейротрансмиттеров (серотонин и др.) [1–4].
Фенилкетонурия I (классическая или тяжелая) — аутосомно-рецессивное заболевание, вызванное мутацией гена фенилаланингидроксилазы (длинное плечо хромосомы 12); выявлены 12 различных гаплотипов, из которых около 90% ФКУ ассоциировано с четырьмя гаплотипами. Наиболее частые мутации в гене фенилаланингидроксилазы: R408W, R261Q, IVS10 nt 546, Y414C. В основе болезни — дефицит фенилаланин-4-гидроксилазы, обеспечивающей конверсию фенилаланина в тирозин, что приводит к накоплению в тканях и физиологических жидкостях фенилаланина и его метаболитов [1–4].
Особую группу составляют атипичные варианты ФКУ, при которых клиническая картина напоминает классическую форму болезни, но по показателям развития, несмотря на проведение диетотерапии, не отмечается положительной динамики. Эти варианты ФКУ связаны с дефицитом тетрагидроптерина, дегидроптеринредуктазы, 6-пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и т. д. [1–4].
Фенилкетонурия II (атипичная) — аутосомно-рецессивное заболевание, при котором генный дефект локализуется в коротком плече хромосомы 4 (участок 4р15.3), характеризующееся недостаточностью дегидроптеринредуктазы, приводящей к нарушению восстановления активной формы тетрагидробиоптерина (кофактор в гидроксилировании фенилаланина, тирозина и триптофана) в сочетании со снижением в сыворотке крови и спинномозговой жидкости фолатов. Результатом являются метаболические блоки в механизмах превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового рядов (L-дофа, 5-окситриптофан). Болезнь описана в 1974 г. [1–4].
Фенилкетонурия III (атипичная) — аутосомно-рецессивное заболевание, связанное с недостаточностью 6-пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптеринтрифосфата (описано в 1978 г.). Дефицит тетрагидробиоптерина приводит к расстройствам, сходным с нарушениями при ФКУ II [1–4].
Примаптеринурия — атипичная ФКУ у детей с легкой гиперфенилаланинемией, у которых в моче в больших количествах присутствует примаптерин и некоторые его производные при наличии нормальной концентрации в спинномозговой жидкости нейромедиаторных метаболитов (гомованилиновой и 5-оксииндолуксусной кислот). Энзиматический дефект пока не выявлен [1–4].
Материнская ФКУ — заболевание, сопровождающееся снижением уровня интеллекта (до умственной отсталости) среди потомства женщин, страдающих ФКУ и не получающих специализированную диету в совершеннолетнем возрасте. Патогенез материнской ФКУ детально не изучен, но предполагается ведущая роль хронической интоксикации плода фенилаланином и продуктами его аномального метаболизма [1–4].
R. Koch и соавт. (2008) при аутопсии головного мозга младенца, у матери которого отмечалась ФКУ (без адекватного контроля за уровнем фенилаланина в крови), обнаружили ряд патологических изменений: низкий вес мозга, вентикуломегалию, гипоплазию белого вещества и задержку миелинизации (без признаков астроцитоза); хронических изменений в сером веществе головного мозга не было обнаружено. Предполагается, что нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита при материнской ФКУ [5].
В практических целях в медико-генетических центрах РФ используется условная классификация ФКУ, основанная на уровнях содержания фенилаланина в сыворотке крови: классическая (тяжелая или типичная) — уровень фенилаланина выше 20 мг% (1200 мкмоль/л); средняя — 10,1–20 мг% (600–1200 мкмоль/л), а также уровень фенилаланина 8,1–10 мг%, если он устойчив на фоне физиологической нормы потребления белка в рационе питания; легкая (гиперфенилаланинемия, не требующая лечения) — уровень фенилаланина до 8 мг% (480 мкмоль/л) [2].
Клинические проявления и диагностика
При рождении дети с ФКУ I выглядят здоровыми, хотя чаще имеется специфический хабитус (светлые волосы, голубые глаза, суховатая кожа). При отсутствии своевременного выявления и лечения болезни в течение первых двух месяцев жизни у них появляется частая и интенсивная рвота и повышенная раздражительность. Между 4 и 9 месяцами становится очевидным выраженное отставание в психомоторном развитии [1–4].
Пациентов отличает специфический («мышиный») запах кожных покровов. Выраженные неврологические нарушения у них редки, но характерны черты гиперактивности и расстройств аутистического спектра. При отсутствии своевременного лечения уровень IQ составляет
В. М. Студеникин, доктор медицинских наук, профессор
Т. Э. Боровик, доктор медицинских наук, профессор
Т. В. Бушуева, кандидат медицинских наук
НЦЗД РАМН, Москва
Что такое фенилкетонурия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Алексенцева Е. С., врача УЗИ со стажем в 12 лет.

Определение болезни. Причины заболевания
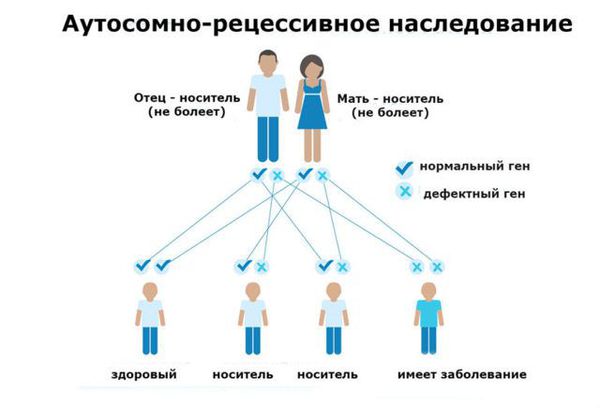
Фенилкетонурия (ФКУ) — генетическое заболевание, в основе которого лежит врождённое нарушение метаболизма аминокислот, характеризующееся повышенным содержанием фенилаланина в крови. Это аутосомно-рецессивная патология, т. е. ребёнок может унаследовать данное заболевание только в том случае, если оба родителя являются носителями дефектной версии гена.
Распространённость фенилкетонурии
Факторы риска фенилкетонурии
Основной фактор риска фенилкетонурии — это наличие у обоих родителей дефекта в гене PAH (Phenylalanine hydroxylase gene). Заболевание развивается, если оба родителя передают ребёнку копию повреждённого гена.
Симптомы фенилкетонурии
Как проявляется фенилкетонурия у новорождённых
Чем дольше ребёнок с фенилкетонурией не получает специфического лечения, тем быстрее развивается умственная отсталость и необратимые нарушения развития. Кроме того, для детей с фенилкетонурией характерны следующие признаки [4] :
Патогенез фенилкетонурии
Фенилкетонурия как самостоятельное заболевание было открыто норвежским врачом Иваром Асбьёрном Фёллингом ещё в 1934 году. Несмотря на это, вопрос о патогенезе долгое время оставался открытым.
Фенилаланин — это незаменимая аминокислота, которая участвует с синтезе белков. Незаменимая она потому, что организм не может самостоятельно её синтезировать, фенилаланин можно получить исключительно из пищи (мясных и рыбных продуктов, творога, сыра, яйц, орехов, хлебобулочных изделий, круп) или с помощью протеолиза — процесса гидролиза белков с помощью ферментов-протеаз.
У пациентов, страдающих фенилкетонурией, из-за дефекта гена и недостатка фермента фенилаланин-гидроксилазы происходит увеличение в плазме крови концентрации фенилаланина (более 1200 мкмоль/л при норме 0-120 мкмоль/л ) и его метаболитов. Одновременно с этим снижается уровень тирозина и его производных (дофамина, адреналина, норадреналина и меланина). Такое состояние оказывает выраженное нейротоксическое действие на структуры мозга. Если пациент с фенилкетонурией не получает или не соблюдает лечение, то у него отмечаются повреждения мозолистого тела, полосатого тела, изменения коры и гипомиелинизация (снижение содержания миелина, образующего оболочку нервных волокон, в различных структурах оболочек мозга). Эти изменения могут привести к снижению интеллектуального развития и нейродегенерации — прогрессирующей гибели нервных клеток. Поэтому пациенты с фенилкетонурией более восприимчивы к нарушениям, связанным с дефицитом дофамина в головном мозге, таким как паркинсонизм.
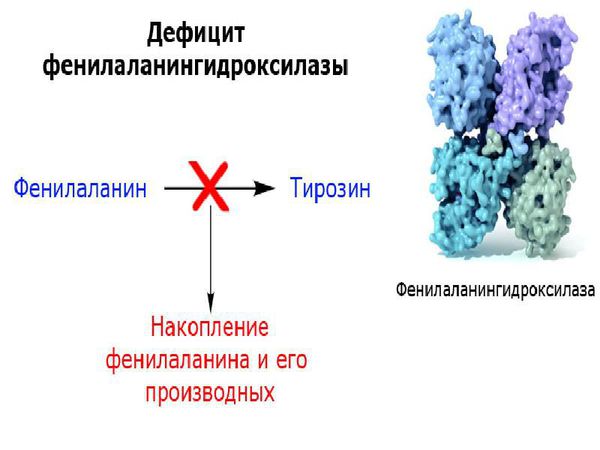
Хотя патофизиологические механизмы повреждения головного мозга у пациентов с фенилкетонурией ещё не совсем понятны, существует множество свидетельств метаболических изменений, которые включают:
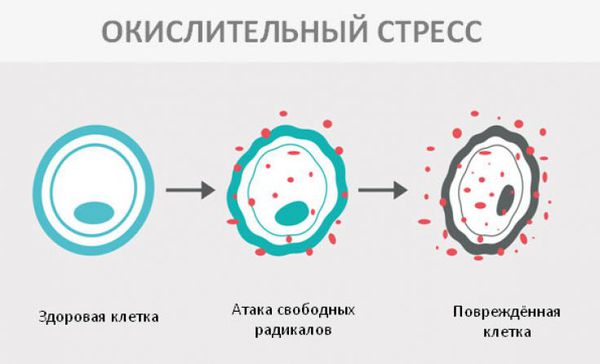
Окислительный стресс — это повреждение клеток активными формами кислорода, которые представляют собой молекулы с повышенной реактивностью из-за наличия неспаренного электрона на внешнем электронном уровне. Активные формы кислорода образуются в клетках постоянно, но в норме их уровень настолько низкий, что организм самостоятельно их нейтрализует с помощью антиоксидантной системы. Окислительный стресс происходит в том случае, когда активных форм кислорода образуется слишком много и антиоксиданты не могут полностью их инактивировать. Такой дисбаланс может вызвать окислительное повреждение белков, липидов или ДНК.
Классификация и стадии развития фенилкетонурии
Пожизненная диетотерапия ассоциирована с нарушением роста, снижением минеральной плотностей костей и дефицитом питательных веществ, что требует постоянного контроля у профильных специалистов.
Диагностика фенилкетонурии
Развитие медицины привело к созданию тандемной масс-спектрометрии для быстрого определения концентраций аминокислот в небольших объёмах крови или плазмы. Данный метод даёт более низкую частоту ложноположительных результатов, измеряя уровни фенилаланина и тирозина в исследуемых образцах.
Для неонатального скрининга медицинским персоналом с помощью скарификатора осуществляется забор крови из пятки новорождённого строго через 3 часа после кормления. Полученные образцы крови помещаются на специальные фильтровальные бумажные тест-бланки и отправляются в лабораторию.
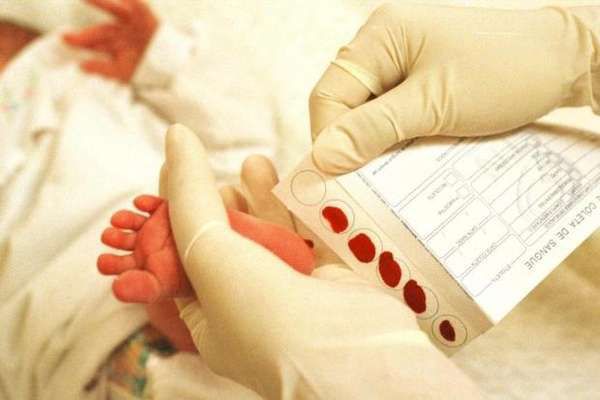
Стоит отметить, что у некоторых детей, особенно у рождённых раньше срока, может наблюдаться незрелость ферментных систем, участвующих в метаболизме аминокислот. Это приводит к кратковременному повышению фенилаланина и положительному результату при скрининге [12] Около 2 % всех случаев повышенного уровня фенилаланина в крови, выявленного при скрининге новорождённых, связаны с нарушением метаболизма кофермента BH4, который участвует в преобразовании фенилаланина. Это подчёркивает важность проведения дифференциальной диагностики при всех выявленных уровнях гиперфенилаланинемии. Фенилкеторурию необходимо дифференцировать с такими заболеваниями, как транзиторная гиперфенилаланинемия недоношенных, наследственная доброкачественная гиперфенилаланинемия, тирозинемия, галактоземия.
Лечение фенилкетонурии
Контроль уровня фенилаланина в крови
Мнение медицинского сообщества относительно начала лечения пациентов с концентрациями фенилаланина 360-600 мкмоль/л достаточно противоречивы. Costello и соавторы [18] проводили исследование, в котором пациенты были разделены на три группы:
Общее потребление белка должно обеспечивать безопасные уровни потребностей данного макронутриента с дополнительной дотацией 40 % L-аминокислот (20 % L-аминокислот необходимы для компенсации потребностей в незаменимой аминокислоте и еще 20 % L-аминокислот используются для контроля фенилаланина в крови).
При введении неадекватных дозировок L-аминокислот они ограничивают синтез белка. Белковый обмен, который в норме складывается из процессов анаболизма (синтеза) и катаболизма (распада) белков, смещается в сторону катаболизма. При данном процессе фенилаланин остается неиспользованным для синтеза белка, и его концентрация в крови будет расти.
При фенилкетонурии необходимо избегать продуктов, богатых белком (мясо, рыба, яйца, обычный хлеб, большинство сыров, орехи и семена), а также продуктов, содержащих аспартам (подсластитель, который используется при изготовлении некоторых газированных изделий и конфет). Пища с низким содержанием белка при диетотерапии данного заболевания должна содержать 50 мг или менее фенилаланина на 100 г сухого продукта. Фрукты и овощи, содержащие менее 75 мг фенилаланина на 100 г пищевого продукта, также могут быть включены в рацион. При употреблении таких овощей, как картофель, брокколи, цветная капуста, брюссельская капуста, нужно учитывать, что даже их небольшое количество в рационе обеспечивает организм 50 мг фенилаланина, поэтому их потребление не должно быть бесконтрольным. Для удобства родителей и ребёнка следует пользоваться «пищевым светофором», где все продукты разделены на группы, разрешённые (ограниченно или неограниченно) и запрещённые к употреблению.
Когда ребёнок маленький, соблюдение диеты не является проблемой для семьи, так как родители контролируют потребление продуктов. В младенческом возрасте предлагается использовать специализированные смеси без добавок фенилаланина, которые дополняются либо грудным молоком, либо стандартной семью для перекрытия суточной потребности в фенилаланине.
Дети более старшего возраста продолжают не только пить специальный безфенилаланиновый продукт, который способен обеспечить потребности в белках и калориях, но и получают дополнительное количество разрешённых продуктов (овощи и фрукты, мёд, животные и растительные масла, зефир, пастила, варенье), необходимых для создания пищевого разнообразия.
По мере взросления ребёнка соблюдение диеты становится всё труднее, так как дети с фенилкетонурией, в отличие от своих сверстников, значительно ограничены в выборе продуктов, что часто приводит к скачкам концентрации фенилаланина у подростков [20] [21] Долгосрочное поддержание диеты необходимо, поскольку пациенты после периода нарушений диеты намного труднее возвращаются к прежнему режиму питания.
Прогноз. Профилактика
Если пациенты с фенилкетонурией получают и соблюдают лечение с самого раннего возраста, то их качество и продолжительность жизни ничем не отличается от их здоровых сверстников.
Пациенты, не получающие или не соблюдающие лечение, часто имеют инвалидность и низкий уровень качества жизни. Кроме того, несоблюдение диеты и отсутствие контроля фенилаланина в организме часто приводит к снижению продуктивности и внимания, нарушению поведения (особенно при уровне аминокислоты свыше 360 мкмоль/л).
Адекватное наблюдение за концентрацией фенилаланина в пределах допустимых показателей достаточно эффективно в профилактике большинства нарушений центральной нервной системы. Большинство людей демонстрируют нормальное общее развитие, легко справляются с образовательными стандартами, ведут самостоятельную жизнь и получают работу, будучи взрослыми.
При соблюдении пациентом режимов диеты и дополнительной дотации минеральных веществ не отмечается увеличения риска таких осложнений, как остеопороз или частые переломы при отсутствии других заболеваний костно-мышечной системы и соединительной ткани.
Профилактика фенилкетонурии
Если у будущих родителей или их близких родственников выявлена фенилкетонурия, то при планировании беременности рекомендуется проконсультироваться с генетиком и пройти обследование. Так можно определить риск рождения ребёнка с фенилкетонурией.
Диагностика фенилкетонурии

Фенилкетонурия (ФКУ) ― наследственное нарушение метаболизма аминокислот, в первую очередь фенилаланина (ФА), входящего в состав белков. Вещество участвует в укладке белка и стабилизации белковых структур.
Первые симптомы: частое срыгивание, рвота, экземы, судороги, исходящий от мочи и кожи запах плесени. Ребенок может был вялым либо, наоборот, гиперактивным. Отстает в психомоторном развитии, наблюдаются признаки олигофрении. Диагноз может быть поставлен в родильном доме. Все дети с фенилкетонурией безусловно получают статус «ребенок-инвалид».
Лечение заболевания заключено в соблюдении специальной низкобелковой диете, не содержащей продукты с ФА.
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.
Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гомонального метаболизмеа и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
Апатичность либо, наоборот, повышенная раздражительность.
Отсутствие интереса к окружению, людям, предметам, обстановке.
Нарушение мышечного тонуса.
Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
Сниженное артериальное давление.
Частое повышение температуры тела.
Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
Гиперсаливация (повышенное слюноотделение).
Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.
Диагностика
Выявить фенилкетонурию можно в первые дни после рождения до появления какой-либо симптоматики. Для определение концентрации фениламина в крови проводят:
Во всех случаях биологическим материалом выступают сухие пятна капиллярной крови младенца.

Если скрининг-тест показал высокие результаты уровня ФА, дополнительно может быть назначено:
Фенилаланин-нагрузочная диагностика для выявления нозологической формы заболевания.
Молекулярно-генетический анализ для установления формы: классическая, II или III тип.
Секвенирование гена РАН, если молекулярно-генетическая диагностика дала отрицательный результат по гену ФАГ.
Анализ на птерины в урине для исключения птерин-зависимых форм.
Дифференциальное диагностирование фенилкетонурии проводят с такими патологиями, как нарушение функции печени, галактоземия и с другими заболеваниями.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Прогноз и профилактика
Проведения массового скрининга в родильных домах позволяет своевременно выявить генетическое отклонение. Вовремя начать соблюдение диетотерапии и, как следствие, предотвратить тяжелые последствия. В противном случае прогноз в отношении умственного развития неблагоприятный.
Классическая ФКУ имеет благоприятный прогноз если диагностирована в первые недели жизни ребенка и соблюдаются все требования врачей. Такие дети ходят в обычные школы, способны получить высшее образовании и вести нормальный образ жизни.

Во время подготовки к беременности пара должна пройти предварительное генетическое тестирование на наличие мутаций в гене РАН. Если у одного из родителей есть дефектный ген, шанс родить ребенка с ФКУ 1:4 и 100% если оба родителя больны.
Женщины с установленной фенилкетонурией при беременности и грудном вскармливании должны соблюдать строгую диету. Высокая концентрация аминокислоты в крови и околоплодных водах оказывает серьезное тератогенное воздействие на плод.
Преимущества АО «СЗЦДМ»
Сдать анализ на уровень ФА можно в подразделениях АО «СЗЦДМ» Здесь вас ждет:
квалифицированных и доброжелательный персонал;
новейшее оборудование, отправка результатов исследований по эл. почте;
несколько вариантов получения данных анализов;
удобное расположение терминалов;
отсутствие очередей, условия конфиденциальности.
Лаборатории находятся в Санкт-Петербурге и других города Ленинградской области, а также в Великом Новгороде, Новгородской обл., Пскове, Калининграде.
Лаборатория АО «СЗЦДМ» предлагает услуги, обеспечивающие комплексное и преемственное лабораторное обследование пациента
Диагностика В медицинских центрах АО «СЗЦДМ» проводят качественные диагностические исследования всего организма
Лечение Наши медицинские центры ориентированы на обслуживание пациентов в амбулаторном режиме и объединены единым подходом к обследованию и лечению пациентов.



