Фенилкетонурия
Фенилкетонурия является генетическим заболеванием, которое связано с нарушением использования организмом одной из важных аминокислот под названием фенилаланин. Основные признаки заболевания – это специфический «мышиный» запах от ребенка, слишком светлая кожа и глаза (альбинизм), а также нарушения в физическом и умственном развитии, общая слабость. Обычно эта болезнь проявляется на первом году жизни ребенка, поскольку нарушения использования фенилаланина проходят очень быстро. Заболевание передается по наследству.
Формы фенилкетонурии
В зависимости от недостатка фермента, который участвует в процессе превращения фенилаланина в тирозин, выделяются следующие формы (каждый из указанных ферментов берет участие в переходе тирозина в фенилаланин, но на различных этапах):
Причины
Фенилкетонурия у детей – это генетический дефект, который передается по наследству.
Симптомы
При такой болезни, как фенилкетонурия, симптомы проявляются уже на первых неделях или месяцах жизни малыша, но только в том случае, если фенилкетонурия не была обнаружена вовремя. Признаки фенилкетонурии:
Диагностика
Диагностика фенилкетонурии проводится в родильном доме на пятый день рождения малыша. С этой целью после кормления берется кровь из пятки ребенка и исследуется уровень фенилаланина. Также возможно исследование мочи. Если уровень аминокислоты высокий, то ребенку назначается специальное лечение. Такие анализы берутся у всех детей.
Если ребенок страдает фенилкетонурией, и обследование не было вовремя проведено, то проявляются характерные признаки, такие как вялость, судороги, бледность кожи и слишком маленький размер головы. В такой ситуации наличие этих признаков и результатов проведенного исследования крови становятся подтверждением наличия заболевания.
Также определить фенилкетонурию можно с помощью генетического анализа, который наиболее часто проводится в случае наличия в семье больного фенилкетонурией. Может потребоваться консультация эндокринолога или медицинского генетика.
Лечение
Диета, предполагающая ограничение потребления фенилаланина, которой должны придерживаться все больные фенилкетонурией. Такой диеты необходимо придерживаться либо до начала полового созревания человека, либо на протяжении всей жизни. Лечение фенилкетонурией включает в себя:
Осложнения и последствия
Если у больного фенилкетонурия, лечение которой было начато своевременно, то прогноз достаточно благоприятный. При правильном соблюдении диеты малыш будет нормально расти и развиваться, а никаких повреждений не будет. В этом случае можно предотвратить опасные последствия в виде нарушений развития головного мозга, разрушений отдельных его клеток – нейронов, а также прочих повреждений. В редких случаях диетотерапия не является эффективной и не помогает избежать неврологических нарушений – судорог, развития умственной неполноценности.
Профилактика фенилкетонурии
Развитие фенилкетонурии не поддается профилактике, поскольку нельзя предотвратить наличие в организме ребенка дефектного гена. Если болезнь развивается, то имеет значение только профилактика появления и развития серьезных повреждений мозга путем правильного и своевременного назначения диетотерапии. С целью пренатальной диагностики (до родов) может быть показано проведение генетического анализа в медико-генетической консультации, что помогает предсказать вероятное развитие заболевания у ребенка. Такие действия проводятся в семьях, где уже были случаи рождения детей с фенилкетонурией.
Диагностика фенилкетонурии

Фенилкетонурия (ФКУ) ― наследственное нарушение метаболизма аминокислот, в первую очередь фенилаланина (ФА), входящего в состав белков. Вещество участвует в укладке белка и стабилизации белковых структур.
Первые симптомы: частое срыгивание, рвота, экземы, судороги, исходящий от мочи и кожи запах плесени. Ребенок может был вялым либо, наоборот, гиперактивным. Отстает в психомоторном развитии, наблюдаются признаки олигофрении. Диагноз может быть поставлен в родильном доме. Все дети с фенилкетонурией безусловно получают статус «ребенок-инвалид».
Лечение заболевания заключено в соблюдении специальной низкобелковой диете, не содержащей продукты с ФА.
Определение заболевания
Фенилкетонурия ― это врожденная, генетическая патология, подразумевающая нарушения гидроксилирования фенилаланина. Характеризуется накоплением в организме аминокислоты и продуктов ее метаболизма, что ведет к тяжелым поражениям центральной нервной системы. Впервые заболевание было описано норвежским врачом И. А. Феллингом в 1934 году.
Изучая болезнь специалисты установили, что за наличие болезни отвечает единственный ген фенилаланингидроксилазы. Первое успешное лечение было разработано и проведено в 1950 году в Англии.

В неонатальном периоде клиника отсутствует. Патология проявляется в первые полгода жизни ребенка. В дальнейшем накопление вещества приводит к тяжелым нарушениям развития. Поэтому крайне важно сразу после рождения выявить дефект и не допустить употребление продуктов, содержащих фенилаланин. Более позднее соблюдение диеты не устранит полученные нарушения, но не допустит развития новых.
Патология одинаково часто встречается среди лиц обоих полов. Расовых особенностей не выявлено. Большое количество больных в таких странах как Китай, Турция, Ирландия. В среднем по России с фенилкетонурией рождается каждый 7-ми тысячный ребенок.
Причины фенилкетонурии
Существует три типа генетического отклонения, первый считается классическим, поскольку диагностируется в более чем 90% случаев. Второй и третий ― более редкая форма патологии. Симптоматика схожа во всех типах, заболевание приводит к умственной отсталости. При классической форме фенилкетонурии избежать этого можно диетотерапией, но атипичные варианты, к сожалению, коррекции не подлежат.
Классическая фенилкетонурия (I тип) ― это низкая выработка фенилаланингидроксилазы (ФАГ), что приводит к собиранию в естественных жидкостях организма фенилаланина и продуктов его расщепления. Патология вызвана мутированным геном РАН.
Фенилкетонурия II типа ― недостаток дигидроптеридинредуктазы, что препятствует преобразованию фенилаланина в тирозин. Патология из-за мутации гена QDPR.
Фенилкетонурия III типа ― недостаток 6-пирувоилтетрагидроптеринсинтазы, нужной для синтеза тетрагидробиоптерина. Патология вызвана мутированным PTS-геном.
Все формы заболевания наследуются по аутосомно-рецессивной форме. Это означает, что генетический дефект может быть унаследован у одного из родителей. Половая принадлежность родителя и ребенка не играет роли.
Классификация
Фенилкетонурия в настоящее время не имеет общепризнанной мировой классификации. Над этим вопросом ведутся дебаты, наравне с изучением заболевания. Чуть ранее, до расшифровки генов, считалось, что степень поражения интеллектуальных способностей зависит от степени активности фермента. Поэтому текущая квалификация признана устаревшей. Не учитывает она и другие симптоматические факторы.
При диагностировании ставят:
I тип (дефицит ФАГ) ― концентрация ФА больше 20 мг/дл.
Средняя форма ФКУ ― ФА от 8,1 до 20 мг/дл.
Легкая форма ГФА-уровень ― ФА от 2,1 до 8,0 мг/дл.
При уровне до 8,0 мг/дл фенилкетонурию считают доброкачественной. Она не требует специального лечения, но необходимо наблюдение первый год жизни ребенка. Контролирует состояние врач-педиатр, невролог, генетик.
Выделяют также еще одну форму фенилкетонурии, не требующую коррекции. Это транзиторная форма ГФА в период новорожденности. Возникает, как правило, при недоношенности, что обусловлено функциональной незрелостью организма. Транзиторная фенилкетонурия ― это временное повышение ФА-уровня, способное подняться до критических значений. При этом клиника отсутствует либо проявления весьма незначительны. Через несколько месяцев биохимические показатели приходя в норму.
Патогенез
Механизм зарождения и развития фенилкетонурии связан с нарушением обмена органического соединения ― аминокислоты фенилаланина. Метаболический блок препятствует преобразованию фенилаланина в тирозин. Аминокислота не только не преобразуется, а накапливается в виде токсичных метаболитов:
фенилэтиламин и прочее.
Скопление фенил-веществ оказывает токсическое действие на ЦНС. В настоящий момент механизм еще до конца не изучен, врачам не известен патогенез дисфункции головного мозга.
Существуют предположения, что поражение нервной системы является результатом ряда факторов. Среди них как прямое токсического воздействие фенилаланина, так и нарушение обмена белков, липопротеидов и гликопротеидов, сбой гомонального метаболизмеа и мембранного транспорта аминокислот. Все это в комплексе имеет важное значение для созревания и правильного функционирования ЦНС.
Симптомы
I тип. Первые признаки у ребенка проявляются в возрасте от 2 месяцев до полугода.
Апатичность либо, наоборот, повышенная раздражительность.
Отсутствие интереса к окружению, людям, предметам, обстановке.
Нарушение мышечного тонуса.
Иногда развивается микроцефалия (малый размер черепа относительно других частей тела) и гидроцефалия (избыточная жидкость, омывающая головной мозг).
К характерным симптомам относятся гипопигментация кожи, волос, радужной оболочки глаз. Моча имеет специфический запах плесени или его еще называют «мышиным» запахом. Эпилептические припадки наблюдаются у половины больных, часто является первым выраженным клиническим симптомом. Приступ характеризуется «салаамовыми» судорогами, напоминающими кивки. Они случаются часто, плохо поддаются антиконвульсантному лечению.
Если не корректировать концентрацию ФА, болезнь прогрессирует. Как правило, уровень IQ у таких детей не превышает 20, при норме от 85. Умственная отсталость настолько сильная, что отсутствуют эмоциональные реакции, наблюдаются психопатии и шизофреноподобные расстройства.
II тип. Первая симптоматика проявляется на первом году жизни.
Сниженное артериальное давление.
Частое повышение температуры тела.
Сухожильная гиперрефлексия (повышение рефлексов) или спастический тетрапарез (обессиливание всех четырех конечностей).
Миоклоническая эпилепсия (генерализованные приступы, преимущественно возникающие после пробуждения).
III тип. Симптомы дефицита пирувоилтетрагидроптеринсинтетазы схож с проявлениями болезни Паркинсона:
Постуральная нестабильность и трудности походки. Сложно либо невозможно поддерживать определенное положение всего тела или конечностей.
Гипокинезия (низкая двигательная активность, ограниченный темп и объем движений).
Гиперсаливация (повышенное слюноотделение).
Окулогирные кризы (симметричное отклонение обоих глаз, обычно направленное вверх).
В 80% случаев этот тип заболевания сопровождается снижением количества биогенных аминов в ликворе. Лечение затруднено тем, что раннее снижение концентрации ФА может вызвать серьезные патологические изменения. Несоблюдение диетотерапии приведет к замедлению развития речи, низкому интеллекту, проблемам с памятью.
Диагностика
Выявить фенилкетонурию можно в первые дни после рождения до появления какой-либо симптоматики. Для определение концентрации фениламина в крови проводят:
Во всех случаях биологическим материалом выступают сухие пятна капиллярной крови младенца.

Если скрининг-тест показал высокие результаты уровня ФА, дополнительно может быть назначено:
Фенилаланин-нагрузочная диагностика для выявления нозологической формы заболевания.
Молекулярно-генетический анализ для установления формы: классическая, II или III тип.
Секвенирование гена РАН, если молекулярно-генетическая диагностика дала отрицательный результат по гену ФАГ.
Анализ на птерины в урине для исключения птерин-зависимых форм.
Дифференциальное диагностирование фенилкетонурии проводят с такими патологиями, как нарушение функции печени, галактоземия и с другими заболеваниями.
Лечение
Симптоматическая терапия при любой формой фенилкетонурии неэффективна. Существует только один способ предотвратить негативные последствия заболевания ― диетотерапия. Из рациона исключают высокобелковые и содержащие фенилаланин продукты. Недостающее количество белка восполняют специализированным лечебным питанием, с максимально низким содержанием аминокислоты ФА или полностью ее лишенным. Следует учитывать, что эффективность терапии напрямую зависит от времени начала коррекции и уже произошедших патологических изменений.
Цель лечебного питания при классической форме заболевания ― это предотвращение развития нарушений ЦНС, физического и умственного развития. Легкая форма ГФА допускает расширение диеты под строгим наблюдением врача за состоянием ребенка и биохимическими показателями. Под запретом: мясо, рыба, орехи, шоколад и бобовые, все виды яиц, творог и сыры. Также следует исключить продукты, содержащие искусственный подсластитель аспартам.
Критерий эффективности лечения ― уровень ФА в крови.
Прогноз и профилактика
Проведения массового скрининга в родильных домах позволяет своевременно выявить генетическое отклонение. Вовремя начать соблюдение диетотерапии и, как следствие, предотвратить тяжелые последствия. В противном случае прогноз в отношении умственного развития неблагоприятный.
Классическая ФКУ имеет благоприятный прогноз если диагностирована в первые недели жизни ребенка и соблюдаются все требования врачей. Такие дети ходят в обычные школы, способны получить высшее образовании и вести нормальный образ жизни.

Во время подготовки к беременности пара должна пройти предварительное генетическое тестирование на наличие мутаций в гене РАН. Если у одного из родителей есть дефектный ген, шанс родить ребенка с ФКУ 1:4 и 100% если оба родителя больны.
Женщины с установленной фенилкетонурией при беременности и грудном вскармливании должны соблюдать строгую диету. Высокая концентрация аминокислоты в крови и околоплодных водах оказывает серьезное тератогенное воздействие на плод.
Преимущества АО «СЗЦДМ»
Сдать анализ на уровень ФА можно в подразделениях АО «СЗЦДМ» Здесь вас ждет:
квалифицированных и доброжелательный персонал;
новейшее оборудование, отправка результатов исследований по эл. почте;
несколько вариантов получения данных анализов;
удобное расположение терминалов;
отсутствие очередей, условия конфиденциальности.
Лаборатории находятся в Санкт-Петербурге и других города Ленинградской области, а также в Великом Новгороде, Новгородской обл., Пскове, Калининграде.
Лаборатория АО «СЗЦДМ» предлагает услуги, обеспечивающие комплексное и преемственное лабораторное обследование пациента
Диагностика В медицинских центрах АО «СЗЦДМ» проводят качественные диагностические исследования всего организма
Лечение Наши медицинские центры ориентированы на обслуживание пациентов в амбулаторном режиме и объединены единым подходом к обследованию и лечению пациентов.
Фенилкетонурия что это такое симптомы у взрослых
Гиперфенилаланинемия (ГФА) представляет собой повышение уровня фенилаланина в крови натощак по сравнению с показателями, выявляемыми у здоровых пациентов идентичного возраста. Гиперфенилаланинемия (ГФА) является группой заболеваний, среди которых наиболее частым является дефицит фенилаланингидроксилазы (ФАГ). Небольшое количество случаев связано с дефектом системы кофактора биоптерина (Burgard et al, 2000).
а) Биохимические и генетические изменения при фенилкетонурии и гиперфенилаланинемии:
1. Дисфункция метаболизма. Для гидроксилирования фенилаланина до образования тирозина необходимо три фермента: ФАГ, карбиноламиндегидротаза (КАД) и дигидропротеинредуктаза (ДПР), и два кофактора: тетрагидробиоптерин (ТГБ) и редуцированный НАД. На основании уровня фенилаланина в плазме и остаточной активности ФАГ в печени выделено три наследственных фенотипа ГФА в связи с дефицитом ФАГ: классическая фенилкетонурия, атипичная фенилкетонурия и нефенилкетонурическая ГФА. Дефекты синтеза и метаболизма 5,6,7,8-тетрагидробиоптерина (ТГБ) являются причиной ГФА и нарушений обмена нейротрансмиттеров.
2. Генетические изменения. ГФА является преобладающим нарушением метаболизма аминокислот среди представителей белой расы, частота заболевания составляет приблизительно 1 на 10000 живых новорожденных. Данное аутосомно-рецессивное заболевание вызвано более чем 500 мутациями локуса ФАГ. Различные группы мутаций преобладают в определенной этнической популяции, что позволяет проводить пренатальную диагностику, выявлять носителей и прогнозировать фенотип фенилкетонурии, связанный с определенным гаплотипом.
Некоторые мутации ФАГ приводят к дефициту ФАГ с остаточной ферментной активностью, которая усиливается под действием ТГБ. В таких случаях фармакологические дозы ТГБ приводят к минимум 30% снижению уровня фенилаланина в крови (Fiori et al., 2005).
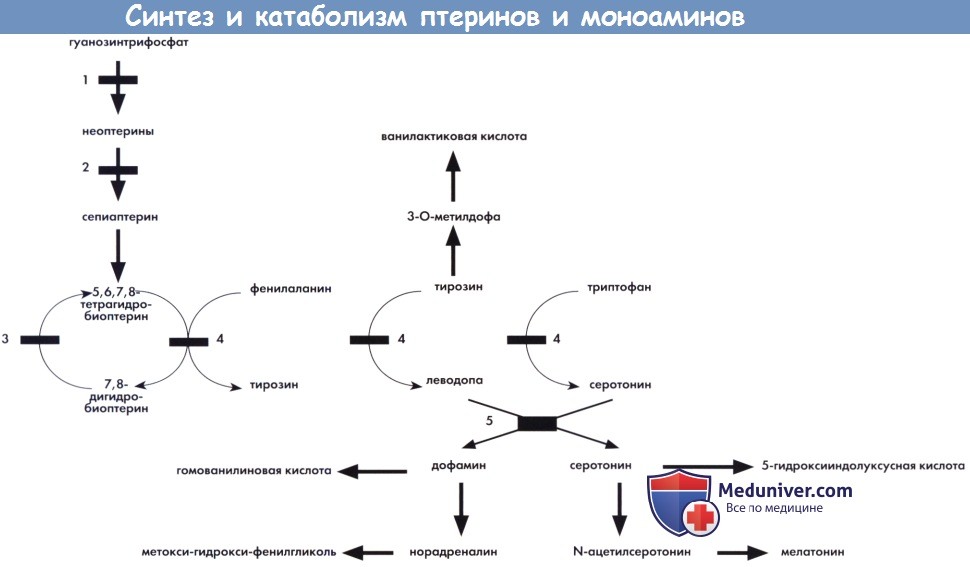 Синтез и катаболизм птеринов и моноаминов
Синтез и катаболизм птеринов и моноаминов
1 — гуанозинтрифосфатциклогидролаза. 2 — биоптеринсинтаза. 3 — дигидроптеридинредуктаза.
4 —фенилаланин-, тирозин-, триптофангидроксилазы.
5 — ароматическая L-декарбоксилаза аминокислот (зависимая от пиридоксальфосфата).
3. Патогенез. Считается, что клинические проявления гиперфенилаланинемии (ГФА) являются результатом накопления фенилаланина и его вторичного воздействия на химические процессы в головном мозге. Фактически, фенилкетонурия чаще всего сопровождается задержкой умственного развития, в то время как при нефенилкетонурической гиперфенилаланинемии умственное развитие не меняется, что предполагает наличие порогового уровня фенилаланина во внеклеточных жидкостях, при превышении которого персистирующая постнатальная (или фетальная) гиперфенилаланинемия приводит к необратимому повреждению головного мозга. В случае если пороговая величина достигается позже, после прекращения соблюдения диеты пациентами, которым ранее проводилось лечение фенилкетонурии, развиваются обратимые химические изменения, способные оказывать воздействие на нейрофизиологические функции.
У пациентов с фенилкетонурией при высоком уровне фенилаланина в плазме отмечается снижение синтеза нейротрансмиттеров. Дефект синтеза нейротрансмиттеров может быть связан с конкурентным ингибированием транспорта крупных аминокислот (тирозина, триптофана и разветвленных цепей аминокислот) в головной мозг через гематоэнцефалический барьер и из спинномозговой жидкости обратно в кровь, что приводит к низкой концентрации тирозина и триптофана в головном мозге пациентов, несмотря на высокое содержание данных веществ в спинномозговой жидкости, а также, возможно, с конкурентным ингибированием гидроксилирования тирозина высоким уровнем фенилаланина.
В головном мозге пациентов старшего возраста, которым не проводилось лечения фенилкетонурии, отмечается аномальная миелинизация, уменьшение массы мозга и снижение содержания миелина. Данное пагубное влияние подтвердилось в ходе исследования на мышах (модель НРН-5). Результаты настоящих или ранее проведенных наблюдений стали основой для гипотезы о снижении количества миелина вследствие ингибирования специфичной для олигодендритных клеток АТФ-сульфурилазы, приводящего к низкому содержанию сульфатидов в миелине, который в свою очередь подвергается протеолитическому распаду. В дальнейшем отмечается потеря нейронов и уменьшение количества межнейронных связей, что было продемонстрировано путем количественной оценки плотности рецепторов нейротрансмиттеров. Если перенести на человека результаты, полученные в ходе исследований на животных, особое поражение гиппокампа и затылочного пространства могут объяснить некоторые нейрофизиологические нарушения у пациентов с фенилкетонурией, которым не проводилось лечения или лечение было недостаточным.
Аномальный синтез белков головного мозга вследствие дезагрегации полисом и снижении скорости удлинения полипептидной цепи могут приводить к снижению массы головного мозга. Дезагрегация полисом отмечается также в сердце и головном мозге плодов крыс, подвергающихся воздействию ГФА беременной самки; данное обстоятельство имеет отношение к фетопатии, связанной с ГФА беременной женщины.
Снижение содержания ДНК и синтеза в нейронах, подвергающихся воздействию фенилаланина в высоких концентрациях, также может объяснять снижение пролиферации нейронов, потерю нейронов и нарушение роста головного мозга.
б) Клинические проявления. Фенилкетонурия при отсутствии лечения Клинические проявления фенилкетонурии, по поводу которой не проводилось лечения, включают задержку умственного развития, неврологические аномалии и вненеврологические симптомы (хотя сроки их возникновения варьируют у различных пациентов). Задержка умственного развития часто сочетается с микроцефалией. Аномалии ЭЭГ встречаются часто (78-95% случаев), но только у 25% пациентов отмечаются припадки, чаще всего генерализованные. Нередко встречается психотическое поведение с гиперактивностью, деструктивным и самодеструктивным поведением, импульсивностью и неконтролируемым поведением с эпизодами эмоционального возбуждения.
У большинства пациентов слабопигментированная кожа с экзематозными проявлениями. Общее физическое развитие обычно хорошее.
Клинический фенотип представляет преимущественно исторический интерес, так как в настоящее время симптомы предотвращаются за счет ранней диагностики и лечения. Тем не менее, до сих пор отмечаются случаи пропущенной ГФА у новорожденных в случае, если тесты не проводились или были получены ложноотрицательные результаты.

в) Фенилкетонурия, по поводу которой проведено раннее лечение. У детей с фенилкетонурией, выявленной при рутинном скрининге новорожденных, по поводу которой вскоре после рождения была начата диетотерапия с ограничением фенилаланина, отмечается нормальный уровень умственного развития (Hanley, 2004). Тем не менее, результаты ретроспективных исследований свидетельствуют о том, что даже при максимально благоприятных условиях лечения у детей отмечается тенденция к более низкому коэффициенту IQ, чем у родственников первой линии, и худшей способности к обучению, чем у сибсов и детей контрольной группы. Часто встречаются субклинические нейрофизиологические (изменения вызванных потенциалов и проводимости нерва) и нейропсихологические нарушения, особенно у пациентов, не строго соблюдающих диету.
Частота аномалий на ЭЭГ увеличивается с возрастом вне зависимости от раннего и строгого соблюдения диеты, а результаты МРТ свидетельствуют о том, что дисмиелинизация является практически универсальным проявлением среди пациентов с классической и атипичными формами фенилкетонурии. Изменения на МРТ затрагивают затылочно-теменные области и в наиболее тяжелых случаях распространяются на лобные и височные доли. Описанные изменения не имеют явной взаимосвязи ни с клиническими или нейропсихологическими проявлениями, ни с контролем потребления фенилаланина в раннем детском возрасте, но коррелируют с уровнем фенилаланина в крови на момент проведения нейровизуализации и частично обратимы при снижении концентрации фенилаланина в крови.
г) Лечение фенилкетонурии и гиперфенилаланинемии (ГФА). У пациентов с классической и атипичной фенилкетонурией для предотвращения необратимых повреждений головного мозга диета с исключением фенилаланина должна быть начата вскоре после рождения. Ежедневное пероральное применение тетрадигидробиопентина может быть альтернативой диете у пациентов с чувствительной к тетрадигидробиопентину формой атипичной фенилкетонурии (Muntau et al., 2002). Для предотвращения интеллектуальной, неврологической и нейрофизиологической деградации после расширения диеты универсальной тактикой является пожизненное лечение. У пациентов с нефенилкетонурической ГФА (уровень фенилаланина в сыворотке
Редактор: Искандер Милевски. Дата публикации: 12.12.2018
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.



